Aanbevelingen
Expert opinion
Achtergrond
Zeer recent is de 5e editie van de WHO Classification of Tumours, Haematolymphoid Tumours verschenen.4 Voorgaande edities kwamen voort uit een samenwerking tussen de WHO, de Society for Hematopathology (SH) en de European Association for Haematopathology (EAHP) ondersteund door adviezen van een klinische adviescommissie (CAC) die uit pathologen, hematologen, oncologen en genetici bestond. De WHO koos er nu echter voor om zelfstandig een nieuwe classificatie vorm te geven zonder hulp van een CAC, hetgeen de SH en EAHP deed besluiten dan maar zonder de WHO een eigen CAC samen te stellen die de WHO classificatie van 2016 moest updaten, leidend tot een zogenoemde International Consensus Classification (ICC).5
Naast vrij essentiële verschillen in andere myeloïde maligniteiten tussen de ICC en de nieuwe WHO classificatie is ook de indeling van CML in fasen duidelijk verschillend. De diagnostische WHO-criteria voor CML in het algemeen staan vermeld in Tabel 1. De WHO heeft de term acceleratiefase vervangen door “hoog-risico chronische fase” (zie Tabel 2 voor de diagnostische criteria), terwijl de ICC de naamgeving acceleratiefase heeft laten bestaan. Het hoofdstuk CML in de WHO classificatie is echter wel door een aantal clinici geschreven die ook deelnemen aan de richtlijnencommissie van het European Leukemia Net (ELN), waarin ook HOVON vertegenwoordigd is. Vandaar dat de HOVON CML richtlijncommissie het hier logischer vindt om de WHO classificatie te volgen.



CML met hoog-risico kenmerken
De reden voor de veranderde indeling van de CML fases is dat in de meeste gevallen een chronische fase met hoogrisicokenmerken (verder “CML in hoog-risico chronische fase” genoemd) nog steeds uitstekend kan reageren op behandeling met een tyrosinekinaseremmer.
Bij 5-10 % van de patiënten wordt bij diagnose niet een standaard translocatie tussen de chromosomen 9 en 22 gevonden, maar zijn meerdere (tot wel 5) chromosomen betrokken.6,7 In die gevallen is er altijd wel een BCR::ABL-fusie en zal bij FISH-onderzoek een normaal fusiesignaal aangetroffen worden. Dergelijke 3-, 4- of 5-wegtranslocaties hebben geen betekenis voor de prognose en er is normale activiteit van de TKIs te verwachten.6,8-14
Patiënten kunnen echter naast de al dan niet complexe translocatie tussen 9 en 22 nog andere afwijkingen hebben in de Philadelphia-chromosoom-positieve cellen, de zogenoemde additionele chromosomale afwijkingen (ACA’s), die de kans op progressie van de ziekte beïnvloeden. Hierbij wordt onderscheid gemaakt tussen hoog-risico ACAs en laag-risico ACAs. De hoog-risico ACAs betreffen: trisomie 8, trisomie 19, deletie 7/del(7q), verdubbeling van het Philadelphia chromosoom, isochromosoom 17q, afwijkingen aan 3q26.2 (waar het EVI1 locus zich bevindt), trisomie 21, 11q23 en complexe aberrante karyotypes.15-17
Indien chromosomaal onderzoek gebruikt wordt voor responsmonitoring kunnen bij 9% van de patiënten ook afwijkingen gezien worden in de Philadelphia-negatieve cellen.18 Het betreft soms uitgebreide afwijkingen. Hoewel in de meeste gevallen de additionele chromosomale afwijkingen voorbijgaand blijken te zijn, is recent gerapporteerd dat patiënten met dergelijke afwijkingen toch een slechtere overleving hadden dan andere patiënten met een 5-jaars kans op progressie van de ziekte van 24% versus 6% en een 5-jaars overleving van 79% versus 94%.19
De aanwezigheid van clonale cytogenetische afwijkingen in Ph-negatieve klonen gaat ook gepaard met een hoger risico op hematologische toxiciteit tijdens TKI behandeling.16
Naast chromosomale afwijkingen is bij een deel van de patiënten sprake van mutaties zoals ook gezien kunnen worden bij clonale hematopoiëse van onbekende betekenis. Bij CML blijkt uit nog vrij beperkte gegevens dat patiënten met deze mutaties, waaronder ASXL1 (voorkomend bij 14-24% van de patiënten bij diagnose) in het algemeen minder goed reageren op de behandeling en minder kans hebben op een succesvolle stop van de behandeling.20-23
Ook tijdens behandeling kunnen kenmerken van hoog-risico ziekte ontstaan. Deze betreffen:
In Tabel 3 staan voor de volledigheid de ICC criteria voor CML-acceleratiefase en blastenfase opgesomd.
CML blastencrisis/blastenfase
Meestal presenteert de ziekte zich in de chronische fase, al of niet met hoog-risicokenmerken, maar bij <1% van de patiënten is de CML reeds in blastencrisis ten tijde van het stellen van de diagnose. Conform de WHO is er bij 20% of meer blasten in bloed of beenmerg sprake van een blastenfase. Dit komt overeen met de tot voorheen gehanteerde ziektedefinitie voor acute myeloïde leukemie: bij een percentage blasten ≥20% wordt gesproken van een blastencrisis, terwijl dit voorheen volgens de ELN pas bij ≥30% het geval was.25
Van speciaal belang is de bevinding van lymfoblasten in het perifere bloed of beenmerg. Volgens de WHO is een toename van het aantal lymfoblasten in bloed of beenmerg een criterium voor blastenfase CML, waarbij opgemerkt wordt dat het exacte percentage waarbij dat geldt onduidelijk is.4 De ICC noemt een percentage van >5% waarbij een blastenfase overwogen moet worden.5
In Tabel 4 staan de volledige WHO 2022-criteria voor de blastencrisis/blastenfase vermeld.
De leukemiewerkgroep geeft de voorkeur aan de WHO 2022-criteria voor de diagnose van CML in chronische fase of blastenfase, omdat recente grote en huidige lopende klinische studies deze criteria hebben aangehouden. De separate acceleratiefase wordt daarmee vervangen door chronische fase met hoog-risicokenmerken (Zie Tabel 2 en 4).4
Op basis van de voorgenoemde gegevens wordt de diagnose “Chronische myeloïde leukemie (CML) BCR::ABL1+” conform de WHO–classificatie van 2022 definitief gesteld en vastgesteld of de ziekte zich in chronische- of blastenfase (“blastencrisis”) bevindt (zie Tabel 2 en 4).
De richtlijncommissie adviseert, in afwachting van verder onderzoek en conform de ICC, een grens van >5% voor het percentage lymfoblasten in bloed of beenmerg als criterium voor blastenfase CML aan te houden.
Nederlands onderzoek heeft laten zien dat patiënten die volgens de vorige WHO classificatie als acceleratiefase werden geduid vanwege een blastenpercentage van 10-15 een even goede uitkomst hebben als patiënten met een lager blastenpercentage.26
De beste voorspeller van CML-gerelateerde dood van patiënten met CML in chronische fase die behandeld worden met een TKI is de ELTS-score.27 De berekening van de ELTS score is conform Tabel 5.
De richtlijncommissie is daarom van mening dat bij alle patiënten met een nieuw geconstateerde CML de ELTS score dient te worden berekend, zodat daarop therapeutische beslissingen kunnen worden gebaseerd. Ook wanneer besloten wordt geen risicogestuurde behandeling in te stellen, is de ELTS score van belang vanwege de prognostische waarde voor het bereiken van een goede moleculaire respons. Daarmee kan een hoge ELTS score leiden tot meer intensieve responsmonitoring.
Routinematig controleren van het cytogenetisch onderzoek van het beenmerg zal in het algemeen niet bijdragen aan aanpassingen in het gevoerde therapeutische beleid en wordt daarom niet geadviseerd.

BCR::ABL naamgeving
Vanwege verschillende redenen is door het HUGO Gene Nomenclature Committee voorgesteld om genfusies in het vervolg aan te geven door de naam van het 1e gen (in cursief en hoofdletters) gevolgd door een dubbele “dubbele punt” (::) gevolgd door de afkorting van het 2e gen.28 Dat betekent dat het BCR::ABL wordt in plaats van BCR-ABL of BCR/ABL. De achtergronden van deze wijziging worden in het gerefereerde artikel besproken en het voert voor deze richtlijn te ver om er hier op in te gaan.
Indeling TKI generaties
Ter verduidelijking volgen hier de definities van de verschillende generaties tyrosinekinaseremmers die beschikbaar zijn voor de behandeling van CML. Imatinib is de eerst geïntroduceerde TKI voor CML. Dit is de enige eerste generatie TKI. Het middel is vrij gevoelig voor een groot aantal mogelijke mutaties in en om de ATP bindingsplaats, waardoor het zijn activiteit verliest. Tweedegeneratie TKIs, zoals dasatinib, nilotinib en bosutinib (in het buitenland ook nog radotinib) zijn minder gevoelig voor de mutaties die imatinib onwerkzaam maken, maar blijven inactief tegen de meest voorkomende T315I mutatie. Ponatinib is een derdegeneratiemiddel, dat ook werkzaamheid behoudt tegen de T315I mutatie. Het werkingmechanisme van asciminib, een vierdegeneratiemiddel, is binding aan de myristoyl pocket, die relatief ver verwijderd is van de ATP bindingsplaats. Daarmee behoudt het middel werkzaamheid tegen vrijwel alle mutaties die conventionele TKIs inactiveren, maar mutaties in en om de myristoyl pocket kunnen de activiteit van het middel verminderen. Asciminib is meestal ook actief in aanwezigheid van de T315I mutatie.
Aanbevelingen voor diagnostiek bij diagnose
De richtlijncommissie adviseert in aanvulling op de kenmerken van hoog risico CML, zoals vermeld in tabel 2, in geval van ASXL1 mutaties, patiënten ook te beschouwen als behorende tot de hoog-risico groep.
Aanbevelingen
Achtergrond
Nederlands onderzoek toonde aan dat Hydrea voorbehandeling geen meerwaarde heeft bij de behandeling van CML en dit wordt daarom in het algemeen afgeraden, behalve bij klachten veroorzaakt door hyperleukocytose of door splenomegalie.
Bij de beslissing welke strategie voor de behandeling met TKIs van nieuw gediagnosticeerde CML te volgen en welk middel te kiezen kunnen de volgende factoren worden meegenomen: biologische hoogrisicofactoren, individuele voorkeuren en wensen van de patiënt op basis van risico op bijwerkingen, huidig cardiovasculair risicoprofiel, leefpatroon, beroep (bijvoorbeeld in ploegendienst), bijkomende medicatie (in verband met interacties) en kosten van de behandeling.
De mogelijkheid om eerder te kunnen stoppen met de behandeling door gebruik van 2G-TKIs kan eveneens meegewogen worden en dit kan bij een zwangerschapswens van groot belang zijn. Alle 2G-TKIs zijn in studies waarin ze vergeleken zijn met imatinib superieur gebleken wat betreft de snelheid en de diepte van de bereikte respons. 29-31 Daarnaast traden er minder progressies op in de 2G-TKI armen van de studies. Zie de overzichtstabellen 7, 8 en 9. Qua overleving is er echter geen verschil tussen imatinib en de verschillende andere TKIs aangetoond.
Patiënten met pre-existente longziekten, perifeer of centraal vaatlijden, of ongecontroleerde risicofactoren daarvoor, worden bij voorkeur met imatinib behandeld.
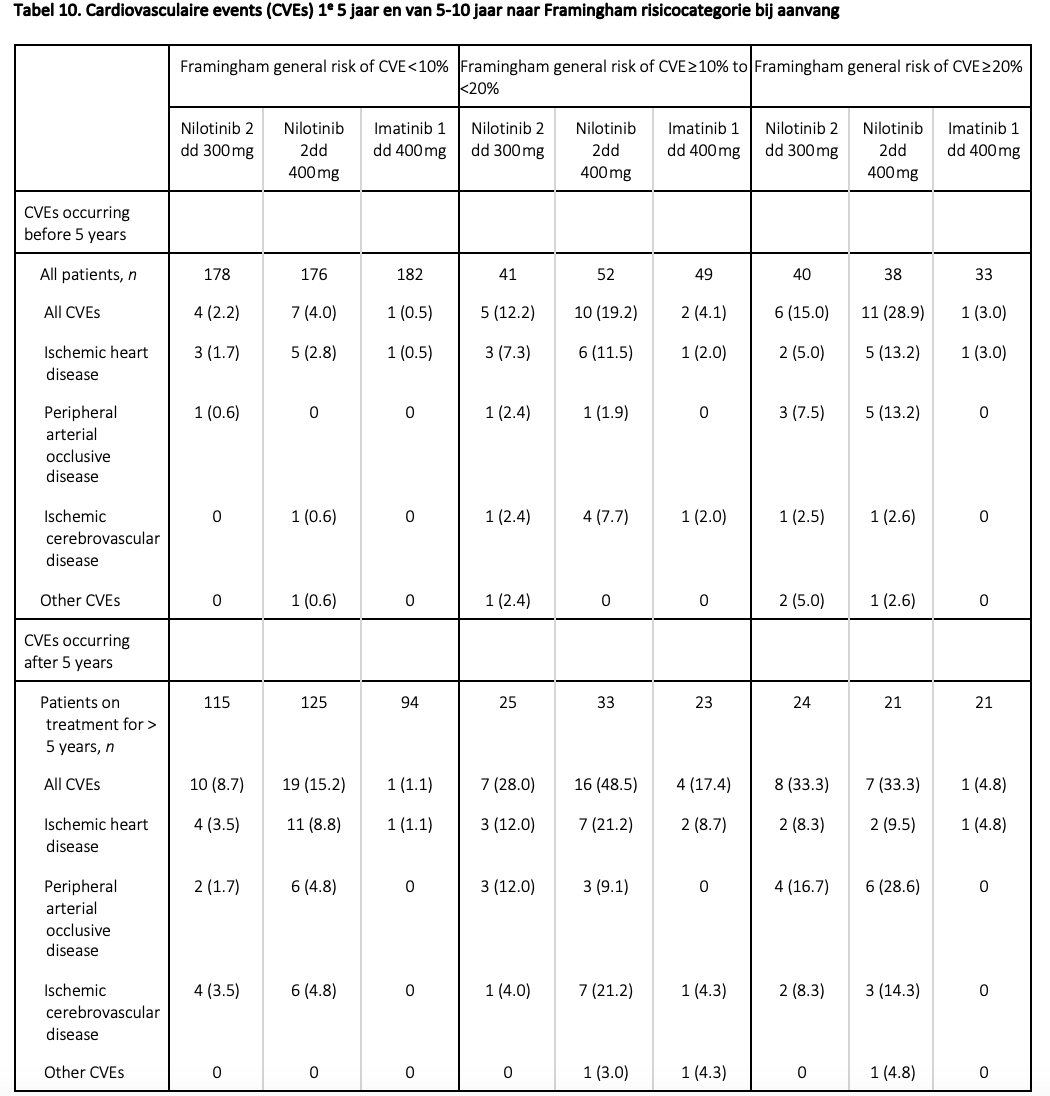
Wanneer dasatinib gegeven wordt, dient men alert te zijn op het ontstaan van pleuravocht en pulmonale arteriële hypertensie (PAH). Gebruik van nilotinib is geassocieerd met irreversibele vasculaire bijwerkingen. Het gaat hierbij om patiënten die myocardinfarcten, CVA’s en perifeer vaatlijden ontwikkelen.29 Daarnaast ontstaat bij 5-6% graad 3-4 hyperglycemie (glucose >13,9 mmol/L) en bij >40% hypercholesterolemie. (tegenover resp. 0% en 7,5% bij imatinib). In de eerstelijns ENESTnd studie werden patiënten met nilotinib (2 dd 300 of 2 dd 400 mg) dan wel met imatinib (1 dd 400 mg) behandeld. De patiënten werden ingedeeld op basis van hun Framingham cardiovasculaire risicoscore. In de eerste 5 jaar na de start van de behandeling was het risico op cardiovasculaire aandoeningen in de twee hoogste risicoklasses (van 3 klassen totaal) resp. 12,2 % en 15% in de nilotinib arm met 2 dd 300mg, en dat was zelfs resp. 19,2% en 28,9% in de 2 dd 400 mg arm.29
In de met imatinib behandelde patiënten waren deze percentages resp. 4,1% en 3,0%. Het verschil in de laagste risicocategorie was beperkter, namelijk 2,2% voor nilotinib 2 dd 300 mg, 4,0% voor nilotinib 2 dd 400 mg en 0,5% voor imatinib.29 Een update met de 10-jaarsdata toont dat ook in de laagrisicogroep duidelijk meer vasculaire complicaties optraden bij gebruik van nilotinib, zie tabel 10.32
Ook bij dasatinib lijkt het cardiovasculair risicoprofiel verhoogd ten opzichte van imatinib (zie tabel 11), maar de gegevens zijn wat meer uiteenlopend.30,33 In een meta-analyse waarin gesuggereerd wordt dat de vasculaire bijeffecten van dasatinib vrijwel even frequent zouden voorkomen als bij nilotinib, werd ook gebruik gemaakt van artikelen die resultaten van tweedelijnsbehandeling beschreven (na falen of suboptimale responses op imatinib) en tevens van een abstract van een onderzoek dat nooit in een peer-reviewed tijdschrift is geplaatst, waarmee de conclusies van de auteurs betwijfeld kunnen worden.34
Datzelfde geldt voor een andere meta-analyse waarin ook verschillende studies werden verzameld en geconcludeerd werd dat nilotinib en ponatinib een duidelijk verhoogd risico op cardiovasculaire bijeffecten geven (resp 2,8 en 10,6 per 100 patiëntjaren), maar dat dit beperkt is voor dasatinib (1,1 per 100 patiëntjaren tegenover 0,8 in de niet aan TKIs blootgestelde groep) met juist een beschermend effect van imatinib (0,1 per 100 patiëntjaren). Hier werden 29 studies geanalyseerd, maar 23 hiervan betroffen abstracts en 1 van de studies was een tweedelijnsonderzoek. Een groot deel was onderzoek van ”real-world” cohorten.35 Los van de vasculaire effecten hebben ouderen bij gebruik van dasatinib meer kans op hartfalen, pleura- en pericardeffusies.36,37
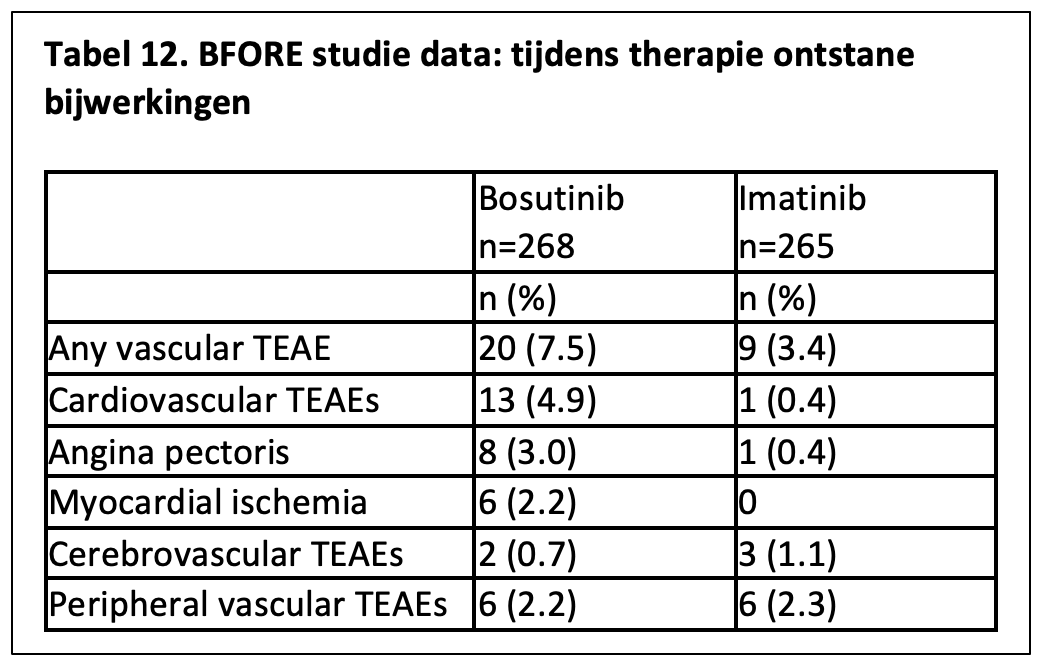
Bosutinib lijkt in de verschillende onderzoeken een relatief mild vasculair risicoprofiel te hebben. In 2 studies, waarin bosutinib in een dosis van 1 dd 500 mg of 1 dd 400 mg vergeleken werd met imatinib 1 dd 400 mg, bleek het percentage cardiovasculaire complicaties (excl. hypertensie) gelijk (4% in 30 maanden) dan wel wat hoger (7,5% na mediaan 55 maanden follow-up) in de bosutinib arm tegenover 3,4% voor de imatinib-arm wat mogelijk verband houdt met een hoger percentage patiënten met cardiovasculaire risicofactoren tussen de studiepopulaties.38,39 (zie tabel 12)
Progressies naar blastenfase (of hoog-risico chronische fase, eerder gedefinieerd als acceleratiefase) komen vooral voor bij de intermediaire en hoog-risico groepen. Een blastenfase heeft ondanks de introductie van diverse zeer krachtige TKIs nog steeds een zeer slechte prognose, die in feite niet verbeterd is sinds de introductie van imatinib.40,41
Behandeling met 2G-TKIs van intermediair en hoogrisicogroepen leidt tot minder gevallen van progressie. (zie tabel 7, 8 en 9) Het risico op progressie naar een late fase was in de ENESTnd studie 2,1-3,5% voor de nilotinib-armen, tegenover 7,4% voor de imatinib-arm, waarbij het verschil vooral bepaald werd door de patiënten in de intermediaire en hoogrisicogroepen.29 Datzelfde geldt voor de Dasision studie en de S0325 studie waarin dasatinib vergeleken werd met imatinib.30,42 Ook gebruik van bosutinib leidde tot minder progressies ten opzichte van imatinib.43 Desondanks is in geen enkele van de uitgevoerde studies een overlevingsvoordeel van de 2G-TKIs voor de gehele groep patiënten aangetoond, los van het genoemde hogere aantal gevallen van progressies in de imatinib-armen van deze studies. (zie tabellen 7, 8 en 9)
De kans op misselijkheid, braken en diarree door bosutinib vermindert wanneer gestart wordt met een dosis van 200-300 mg, welke vervolgens wekelijks wordt opgehoogd tot 1 dd 400 mg.
Tenslotte moet bij elke patient het belang van niet roken worden onderstreept, vanzelfsprekend in het kader van cardiovasculair risicomanagement, maar ook omdat roken de kans op progressie naar blastencrisis verhoogt.44,45
Steeds meer 2e generatie TKIs verliezen hun patentbescherming. Daarmee kan bijv. generiek dasatinib de drempel om een keuze te maken voor een 2G-TKI als eerstelijnstherapie verlagen. Imatinib en dasatinib kosten nu ongeveer 1300,- per jaar, terwijl de andere middelen nog tenminste 36.000,- per jaar kosten. Het patent van bosutinib loopt overigens ook af in september 2024.
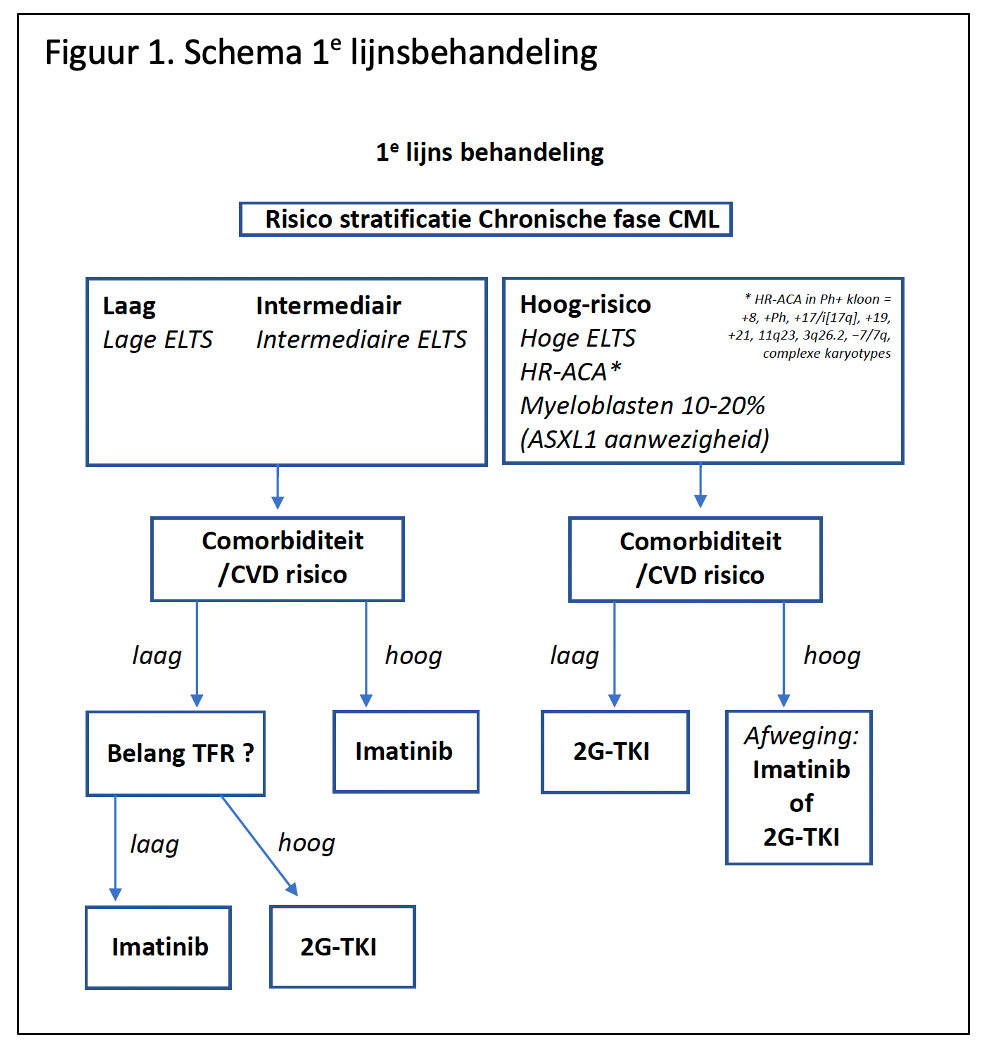
Figuur 1. geeft bovenstaande adviezen schematisch weer.
Aanbevelingen
Achtergrond
In Nederland wordt het BCR-ABL meestal met een Cepheid/GeneXpert machine kwantitatief bepaald. Hierbij wordt, naast het aantal kopieën BCR::ABL, parallel ook het aantal kopieën van een controle-gen meegemeten. Dit betreft bij de GeneXpert ABL, maar in andere methoden wordt soms GUSB gebruikt, hetgeen ook acceptabel is. De resultaten dienen weergegeven te worden in de internationale schaal, die gebaseerd is op een gemiddeld uitgangsniveau van 30 patiënten die aan de IRIS-trial meededen. Weergaves in zogenaamde “logreducties” of vergelijkingen ten opzichte van het initiële diagnosemateriaal van de patiënt zelf geven verwarring en/of zijn onduidelijk en zijn daarom niet acceptabel.
Wanneer in een monster geen BCR::ABL-transcripten meer kunnen worden aangetoond dient de term “complete moleculaire respons” vermeden te worden. In plaats daarvan moet een maat voor de gevoeligheid van de test aan het moleculaire responsniveau toegevoegd worden. Deze gevoeligheid van de test wordt bepaald door het gemeten aantal kopieën van het controle-gen.47
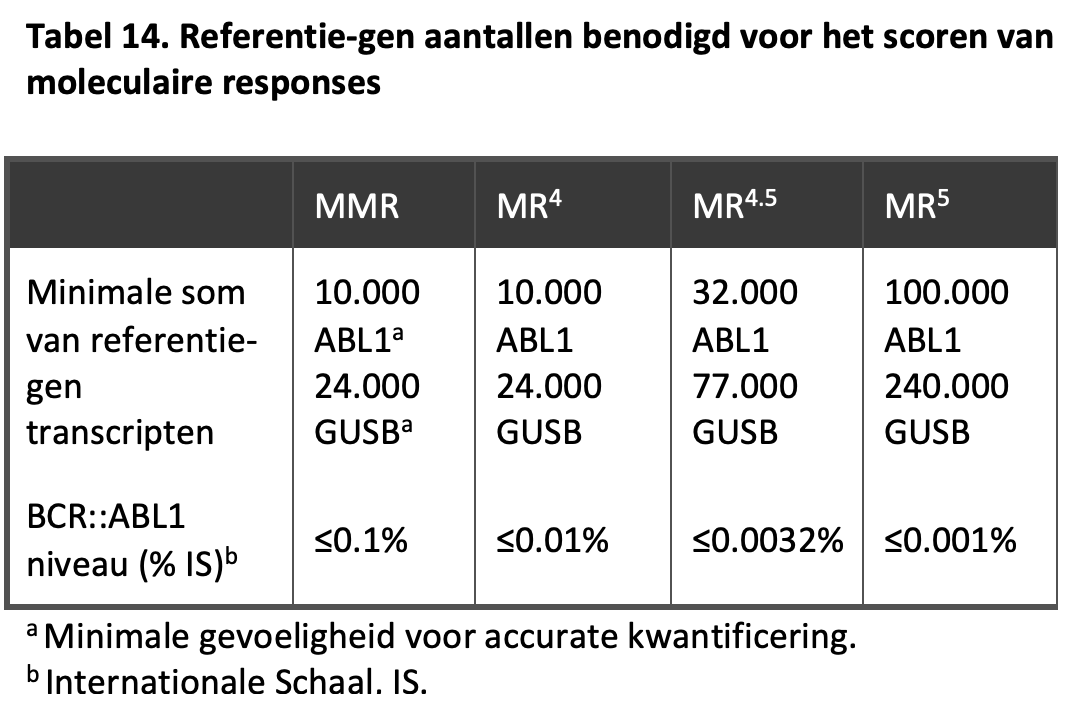
Een major moleculaire respons (MMR) betekent een meetbaar BCR-ABL niveau van 0,1% of lager, een MR4 betekent een meetbaar BCR::ABL-niveau onder 0,01%, òf een onmeetbaar niveau bij tenminste 10.000 ABL-transcripten of tenminste 24.000 GUSB-transcripten. Voor een MR4.5 geldt een meetbare waarde onder 0,0032% of een onmeetbaar niveau met 32.000-99.999 ABL-transcripten of 77.000-239.999 GUSB-transcripten. Een MR5 is gedefinieerd als een meetbaar BCR::ABL-niveau <0,001% of een onmeetbaar niveau met tenminste 100.000 ABL- of 240.000 GUSB-transcripten. Voor gedetailleerde informatie over de geadviseerde rapportage van de BCR::ABL-uitslagen wordt verwezen naar een vrij recente publicatie.47 Zie ook Tabel 14.
Het is van groot belang dat tijdens behandeling regelmatig en frequent het BCR::ABL-niveau gemeten wordt. Geadviseerd wordt dat bij een CML in chronische fase in het eerste jaar van de behandeling tenminste na 3, 6, 9 en 12 maanden te doen. Na een jaar kan de frequentie bij patiënten die een MMR hebben behaald terug naar eens per 4 maanden. Na 3 jaar stabiele MMR is controle elke 6 maanden acceptabel. Dit komt voort uit de zeer geringe kans op progressie als eenmaal een MMR bereikt is.48
Voor patiënten die op de genoemde tijdspunten geen optimale respons hebben behaald wordt geadviseerd 4-6- wekelijkse BCR::ABL-monitoring te verrichten teneinde het resultaat van eventuele aanpassingen van de therapie goed te kunnen controleren dan wel progressieve stijging van het BCR::ABL-niveau tijdig te kunnen onderkennen.
Na de diagnose kan volstaan worden met BCR::ABL-metingen in perifeer bloed.24 Beenmerganalyse wordt geadviseerd bij diagnose, bij aanhoudende graad 3-4 hematologische toxiciteit en in geval van falen van de therapie, teneinde progressie van de ziekte uit te sluiten. Daarnaast kan, voorbij de eerstelijnsbehandeling, een complete cytogenetische respons in het beenmerg in het geval het BCR::ABL-niveau rond 1% blijft fluctueren beschouwd worden als een acceptabele respons en dus als een reden de huidige behandeling niet te veranderen (zie ook volgende sectie).49
Poliklinische consulten kunnen na het bereiken van een stabiele MMR vervangen worden door video- of telefonische consulten. Bij verlies van respons of verdenking op bijwerkingen, zoals PAH of pleuravocht, moet de patiënt alsnog in het ziekenhuis gezien worden.
De commissie acht het van belang dat bij elk consult de “therapietrouw” en eventuele psychosociale gevolgen van de ziekte en/of behandeling met de patiënt besproken kunnen worden.
Aanbevelingen
Achtergrond
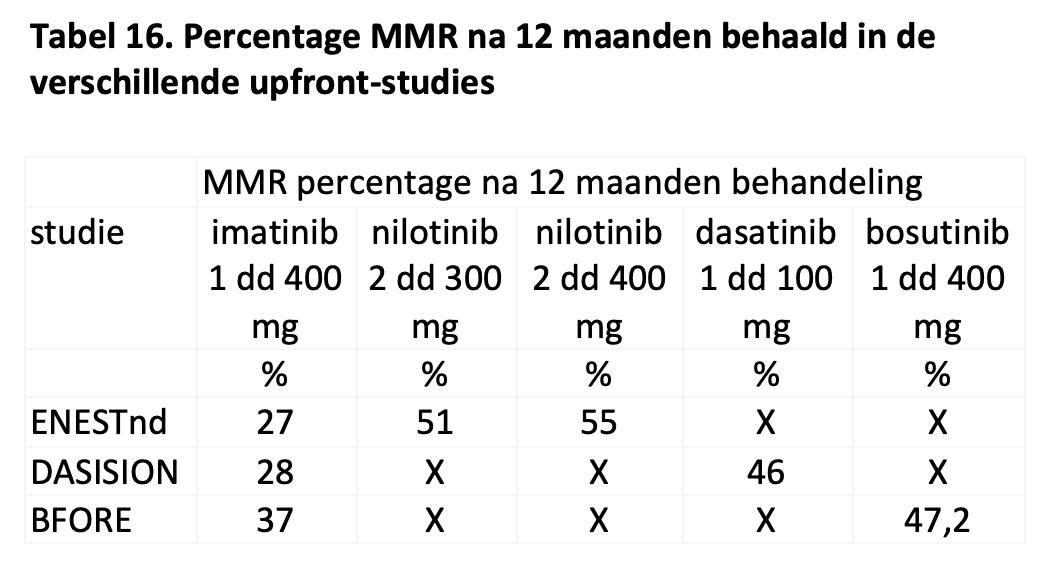
De meerderheid van de patiënten die met imatinib als eerstelijnsbehandeling worden behandeld bereikt geen major moleculaire respons na 1 jaar behandeling en voldoet daarmee niet aan de criteria voor een optimale respons. Met tweedegeneratie TKI’s is het percentage patiënten met een optimale respons beduidend hoger, maar nog altijd behaalt 45 tot 54% niet een BCR::ABL niveau <0,1% na 1 jaar behandeling.30,40,43 (zie Tabel 16) Dat is niet persé een probleem. Immers, een groot deel van de patiënten behaalt later alsnog een MMR. Het is vooral van belang de patiënten te identificeren die falen op de therapie. Dergelijke patiënten dienen overgezet te worden op een ander middel en tevens moet diagnostiek verricht worden om te kunnen begrijpen waarom de patiënt onvoldoende reageert en zo nodig gericht andere therapie in te zetten.
Voor het vaststellen van de oorzaken van onvoldoende respons is als eerste van belang therapietrouw uit te vragen. Ongeveer 25% van de patiënten neemt de voorgeschreven medicatie onvoldoende nauwkeurig in, hetgeen betekent dat meer dan 10% van de tabletten per maand niet wordt ingenomen.50,51 Daarnaast kunnen door interacties met andere geneesmiddelen onvoldoende spiegels worden opgebouwd waardoor het therapie-effect onvoldoende blijft. In beide gevallen kunnen spiegelmetingen uitkomst bieden. Indien subtherapeutische niveaus worden vastgesteld kan ofwel een verhoging van de dosis dan wel een verandering van middel worden gekozen. Bij de keuze van een ander middel vanwege adherentie-problemen is het innameschema van het nieuwe middel van belang. Nilotinib dient bijvoorbeeld tweemaal daags op een lege maag ingenomen te worden en is, naast de cardiovasculaire risico’s, ook daarom een minder geschikt middel dan de andere 2G-TKIs.
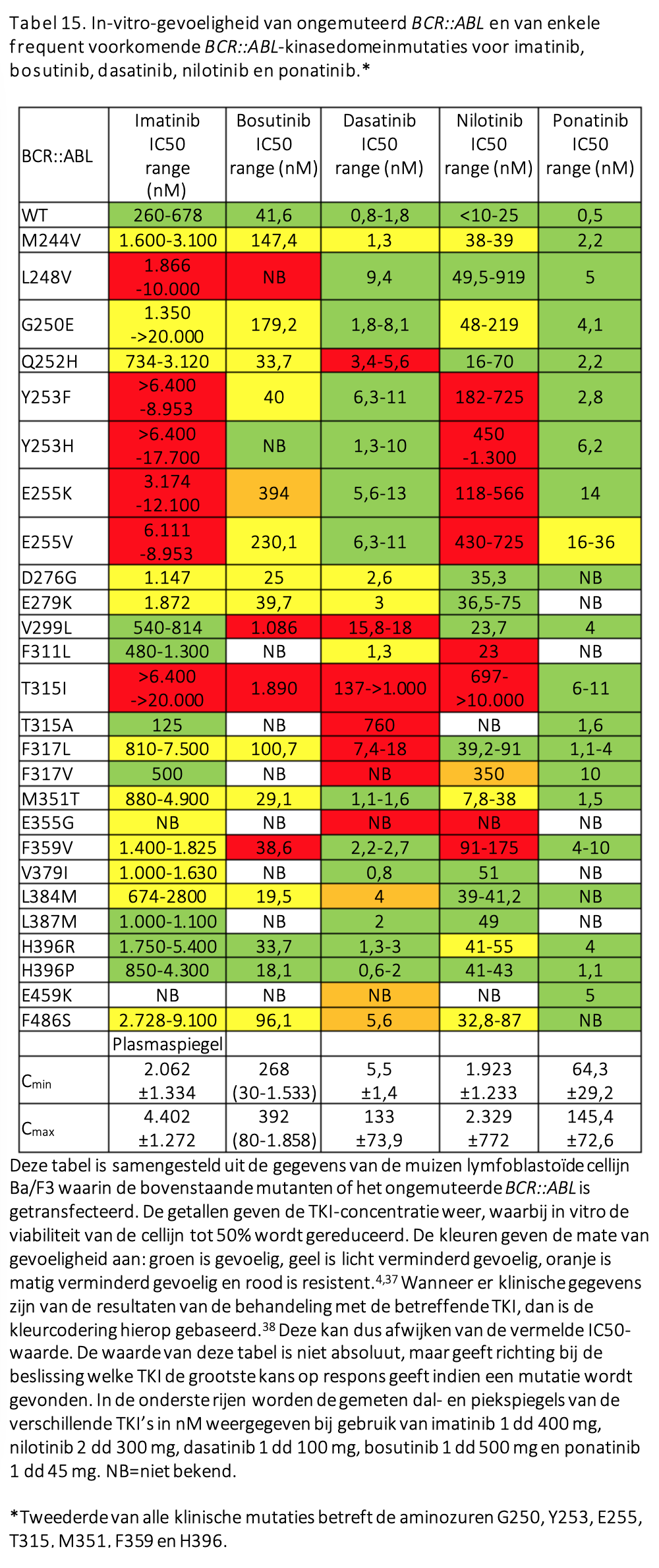
Voor patiënten die resistentie vertonen terwijl de bloedspiegels van het gebruikte middel wel adequaat zijn is aanvullend onderzoek in de vorm van mutatieanalyse van het ABL kinase-gen geïndiceerd. Mutaties die veroorzaken dat verschillende middelen minder goed kunnen binden aan de ATP-bindingspocket in het BCRABL eiwit komen in 25-45% voor bij patiënten die primair resistent zijn voor de eerstelijnsbehandeling, maar bij bijna 60% van de patiënten die in tweede instantie verlies van respons vertonen, dus na een initiële adequate respons (secundaire resistentie).
Mutaties kunnen de keuze van een volgende therapielijn sturen. Sommige mutanten zijn nog wel gevoelig voor dasatinib of bosutinib, dan wel nilotinib, maar de veelvoorkomende T315I mutatie is alleen gevoelig voor ponatinib (en voor asciminib, maar dat middel is niet voor de tweedelijn en niet voor T315I gemuteerde CML geregistreerd, zie verder).
Voor patiënten die intolerant zijn voor de eerstelijnsbehandeling is het advies om na imatinib in eerste instantie een keuze te maken uit dasatinib of bosutinib. Wanneer daarmee niet uitgekomen kan worden of bij duidelijke contra-indicaties tegen deze middelen is nilotinib een alternatief. Zie hiervoor ook de module “Hoe moet omgegaan worden met bijwerkingen van de behandeling?”.
Er kunnen redenen zijn om een stabiel BCR::ABL niveau van rond de 1% te accepteren in de eerste lijn, bijvoorbeeld als een oudere patiënt dit niveau bereikt heeft op imatinib en de 2G-TKIs (relatief) gecontra-indiceerd zijn vanwege, onder meer, cardiovasculaire comorbiditeiten.
Aanbevelingen
Achtergrond
De WHO-criteria voor het vaststellen van een chronische fase met hoog-risicokenmerken of blastencrisis zijn vermeld in Tabel 1.4 Wanneer er bij diagnose reeds een chronische fase met hoog-risicokenmerken bestaat kan het resultaat van behandeling met TKIs uitstekend zijn. Wel wordt geadviseerd extra zorgvuldige responsmonitoring te verrichten met kleinere intervallen dan normaal totdat een optimale respons is bereikt, waarna reguliere monitoring plaats kan vinden. Behandeling met een 2G-TKI, dasatinib of bosutinib, heeft in dergelijke gevallen de voorkeur van de commissie.
Het is zeer ongewoon dat een chronische fase met hoog-risicokenmerken ontstaat tijdens behandeling met TKI als er telkens voldaan is aan de responsmijlpalen. Veelal zal het juist gaan om patiënten die onder verschillende lijnen van therapie onvoldoende respons hebben bereikt. Een zich ontwikkelende chronische fase met hoog-risicokenmerken is dan een sterke waarschuwing voor het ontstaan van een blastencrisis. Inmiddels zal in dergelijke gevallen reeds onderzoek gedaan zijn naar BCR::ABL-mutaties en dit dient bij het vaststellen van de hoog-risicokenmerken zeker opnieuw te gebeuren aangezien sequentiële mutaties geregeld voorkomen, als uiting van genetische instabiliteit van een niet goed onderdrukte CML-kloon (zie Tabel 15).76 Bij het vaststellen van een mutatie die gevoelig is voor een niet eerder gegeven middel, kan behandeling hiermee gestart worden, opnieuw onder strikte responsmonitoring met intervallen van 4-6 weken. Intussen is het bij geschikte patiënten verstandig te gaan zoeken naar een allogene donor, zodat bij uitblijven van een acceptabele respons een stamceltransplantatie verricht kan worden.
Indien er geen mutatie wordt gevonden bij gebruik van imatinib kan een 2G-TKI geprobeerd worden en bij uitblijven van een acceptabele respons nog ponatinib. Als derdelijns behandeling is asciminib ook een goede keus en wellicht te prefereren boven ponatinib wegens de betere tolerantie. Wanneer een acceleratie optreedt onder gebruik van een 2G-TKI dient direct met ponatinib gestart te worden indien geen richtinggevende mutatie gevonden wordt. Het advies een donorsearch te starten geldt vanzelfsprekend ook hier.
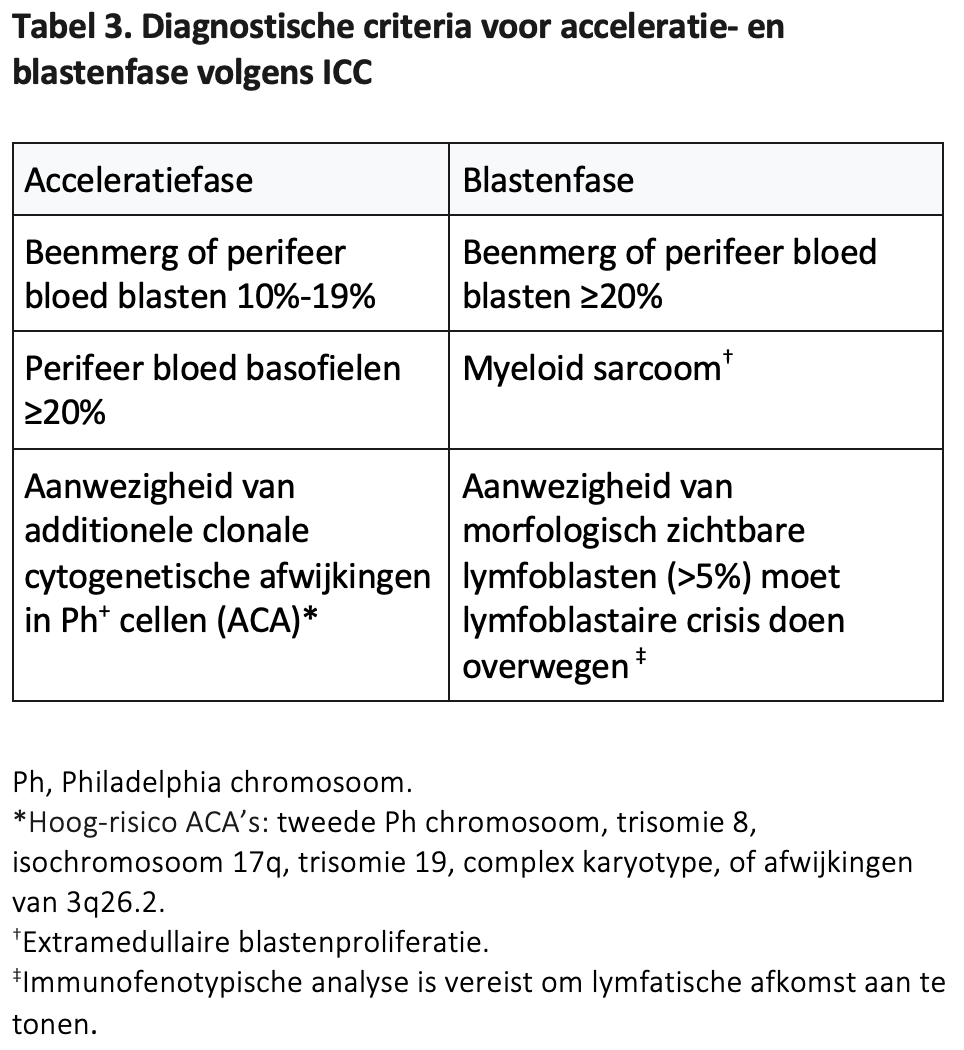
Zelden presenteert zich een CML in blastencrisis.77 Het is dan vaak moeilijk om vast te stellen dat er een chronische fase aan vooraf gegaan is. Als er een regulier p210 transcript bestaat, zoals gezien wordt bij een e13a2 of e14a2 fusie, dan betreft het waarschijnlijk toch een CML, hoewel de WHO in de 2022 update van de classificatie van hematologische neoplasmata ook de entiteit “AML with BCR::ABL1” heeft vastgelegd, daarmee aangevend dat in gevallen waarin een klinisch manifeste chronische voorfase ontbreekt de diagnose CML niet met zekerheid te stellen is.4
Bij deze AML met BCR::ABL1 translocatie worden mutaties of additionele chromosomale afwijkingen zoals vaak voorkomen bij myeloïde blastencrises die uit een chronische fase CML ontstaan minder vaak gezien.4 In geval van twijfel kan het diagnostische algoritme uit Figuur 1 gevolgd worden.78
In de lymfatische blastencrisis voortkomend uit chronische fase CML komen cryptische deleties in immuunglobuline- en T-cel receptorgenen vrijwel altijd voor en gaan dan gepaard met verlies van IKZF1 en/of CDNK2A/B genen zoals met CGH-array kan worden vastgesteld.76
Behandeling met imatinib van een de-novo blastencrisis (BC) CML induceert in 15-27% complete remissies, maar hoewel soms snel optredend zijn ze meestal van korte duur, bij lymfatische BC minder dan 100 (mediaan) en bij myeloïde BC minder dan 200 dagen.79-81 Er zijn geen head-to-head prospectieve vergelijkende studies verricht tussen imatinib en 2G-TKIs bij de behandeling van CML-BC, waardoor er geen hard oordeel te geven is over de beste TKI in deze setting. Wel werd in een retrospectieve serie een betere overleving gevonden bij patiënten die initieel met een 2G-TKI werden behandeld ten opzichte van imatinib.82 Daarnaast induceren dasatinib (en nilotinib) alsnog 35-80% complete hematologische remissies van CML-BC bij eerdere imatinib-resistentie, hoewel deze meestal ook niet duurzaam zijn.83-85 Dasatinib passeert bovendien beter de bloed-hersenbarrière dan imatinib.86,87 De commissie acht daarom initiële behandeling met dasatinib (1 dd 140 mg) voor een nieuw geconstateerde en niet eerder behandelde CML-BC superieur aan die met imatinib. Nilotinib is niet geregistreerd voor behandeling van CML-BC.
Ponatinib is in een fase 2 onderzoek getest in CML-BC en daarbij werd een major hematologisch respons percentage van 31% gezien. De mediane duur tot een MHR was 4,1 weken en bij de 47% van de patiënten die een MCyR bereikten had 66% die nog steeds na 1 jaar. Aangezien dasatinib en ponatinib niet met elkaar in een onderzoek zijn vergeleken kan de commissie geen voorkeur uitspreken voor dasatinib versus ponatinib. Wel is het vanzelfsprekend dat in geval er bij mutatieanalyse een dasatinib-resistente mutatie wordt gevonden, ponatinib wordt gegeven. Bosutinib is ook voor blastencrisis geregistreerd, maar het aantal patiënten dat in studies behandeld is, is beperkter dan voor dasatinib en ponatinib en bovendien was het responsniveau daarbij lager, zodat de commissie bosutinib als tweede keus middel na dasatinib of ponatinib beoordeelt.85,88-90 Van asciminib zijn nog te weinig gegevens bekend om een oordeel op te baseren.
Er zijn geen goede studies verricht om vast te stellen of TKIs wellicht met chemotherapie moeten worden gecombineerd wanneer een BC wordt gediagnosticeerd. Desondanks suggereren verschillende series dat een combinatie van AML gebaseerde behandeling met een TKI bij een myeloïde BC en van ALL gebaseerde chemotherapie in combinatie met een TKI de kans op het bereiken van een complete hematologische respons of complete remissie en het uiteindelijk bereiken van een allogene stamceltransplantatie verhoogt.82 Een recente studie suggereert overigens dat zowel een myeloïde als een lymfatische blastencrisis met een combinatie van FLAG-IDA chemotherapie plus ponatinib kan worden behandeld.91 Dit betreft een klein (n=17) en niet gerandomiseerd onderzoek en daarom is de bewijskracht voor het algemeen gebruik van dit regime, onafhankelijk van de myeloïde of lymfatische origine, beperkt.
Zeker bij patiënten die zich presenteren met zeer hoge leukocytengetallen, dan wel niet snel lijken te reageren op de ingestelde TKI-behandeling, moet daarom combinatietherapie overwogen worden.92,93 De combinatie verhoogt wel de kans op toxiciteit van zowel chemotherapeutica als TKI’s.
Chemotherapie dient ook overwogen te worden in die gevallen waarin het niveau van restziekte te hoog blijft onder TKI-behandeling. Het verrichten van een alloSCT in een complete hematologische remissie leidt tot betere resultaten dan wanneer de blastencrisis nog steeds actief is ten tijde van de alloSCT. Uit een retrospectieve analyse van een groot cohort Chinese patiënten(n=386) lijken diepere responsniveaus voor alloSCT tot betere resultaten te leiden, zowel bij myeloïde als bij lymfatische blastencrises. Moleculair ondetecteerbare ziekte onder behandeling van TKI met of zonder chemotherapie gaf de beste overleving (75% na 5 jaar), hetgeen beter was dan alleen een MMR, hetgeen weer beter was dan een CCyR of CHR, hetgeen weer beter was dan bij het uitblijven van een CHR.94 De commissie beveelt daarom aan tenminste een complete hematologische remissie na te streven ten tijde van de start van de conditionering van de alloSCT. Waarschijnlijk is een complete cytogenetische respons beter en een major moleculaire respons of ondetecteerbaar BCR::ABL een optimale uitgangssituatie voor de start van de alloSCT maar de bewijskracht van deze stelling is laag door de geringe kwaliteit van de weinige studies.95
De rol van blinatumomab in combinatie met dasatinib of ponatinib is wel in Ph+ ALL onderzocht, maar niet in lymfatische CML-BC.96,97 De resultaten van de combinaties in ALL zijn echter opvallend goed en dit suggereert dat ook bij een lymfatische CML-BC een dergelijke combinatie de voorkeur zou verdienen. Of hiermee ook een allogene stamceltransplantatie onnodig zou worden zoals gesuggereerd wordt in de genoemde studies is voor lymfatische CML-BC onduidelijk. Beschikbare gegevens wijzen op hogere persisterende MRD niveaus bij CML-BC dan bij Ph+ ALL onder behandeling met chemotherapievrije combinatietherapie.97 De commissie handhaaft daarom het advies om bij een lymfatische CML-BC te streven naar een alloSCT bij het bereiken van een goede respons.
Bij patiënten die geen AML of ALL-gebaseerde chemotherapie kunnen ondergaan dient de TKI als monotherapie voortgezet te worden tot aan progressie en dat geldt vanzelfsprekend ook voor patiënten die wel intensieve therapie hebben ondergaan, maar niet kwalificeren voor een alloSCT vanwege uiteenlopende redenen.
Bij het vaststellen van het optimale beleid dient de behandelaar zich te realiseren dat de uitkomsten van de behandeling van CML-BC nog immer teleurstellend zijn en dat in bepaalde gevallen palliatie de beste optie is.40,98
Bij patiënten die een BC ontwikkelen onder TKI-behandeling kan de patiënt onder 4-wekelijkse meting van het BCR::ABL-niveau zo lang de transplantatievoorbereiding loopt de volgende generatie TKI continueren.
In alle situaties moet beoordeeld worden of de patiënt met een BC in aanmerking kan komen voor een studie.
Over de conditionering voor de alloSCT is geen op bewijs gebaseerd advies te geven. Bij een chronische fase stelt de werkgroep voor een verminderd intensief schema te hanteren, te meer daar bij een eventueel recidief de kans op het alsnog bereiken van solide remissie onder invloed van donorlymfocyteninfusie zeer groot is.99 Bij een blastencrisis is een myelo-ablatief schema bij daarvoor geschikte patiënten wellicht te prefereren, maar de meerwaarde hiervan boven verminderd intensieve conditionering is onbewezen.
Er is onduidelijkheid over de rol van onderhoudsbehandeling en timing van TKIs ná een allogene stamceltransplantatie. De commissie adviseert bij patiënten met of zonder mutaties die eerder gevoeligheid hebben vertoond voor een al dan niet op de mutatie toegesneden TKI, deze door te zetten gedurende tenminste een jaar na de transplantatie waarbij minstens een jaar geen detecteerbaar BCR::ABL aantoonbaar moet zijn geweest. Wellicht is een langere onderhoudsbehandeling veiliger, maar het ontbreekt aan gegevens in de literatuur. Bij patiënten die ongevoelig bleken voor TKIs vóór de transplantatie is het doorzetten van deze middelen na transplantatie niet rationeel, tenzij de patiënt een mutatie blijkt te hebben waarvoor eerder niet het geschikte middel gegeven is.
Ook bij een blastencrisis dient bij diagnose mutatie-analyse verricht te worden, aangezien mutaties frequent voorkomen (tot >80%) en dit de behandeling kan sturen.100 Bij elk eventueel recidief dient deze analyse opnieuw plaats te vinden als er therapeutische consequenties aan de uitslag te verbinden zouden kunnen zijn.