Hemofilie is een zeldzame, erfelijke bloedingsziekte met een geslachtsgebonden overervingspatroon. Bij ongeveer 90% van de patiënten is er sprake van hemofilie A met een verlaagd of afwijkend stollingsfactor VIII en bij 10% is er sprake van hemofilie B met een verlaagd of afwijkend stollingsfactor IX. Hemofilie wordt ingedeeld in verschillende vormen van ernst, afhankelijk van de hoeveelheid resterend factor VIII of IX: van mild (>5 IE/dL), matig-ernstig (1-5 IE/dL) tot ernstig (<1 IE/dL).
Voor de behandeling van hemofilie zijn verschillende stollingsfactor VIII en IX-concentraten beschikbaar. Deze concentraten zijn ofwel uit donorplasma gezuiverd, dan wel via recombinante DNA-technieken geproduceerd. In recente jaren zijn de recombinante producten op verschillende wijzen gemodificeerd met als voornaamste doel de halfwaardetijd van de stollingsfactor en daarmee de werkingsduur ervan te verlengen.
Suppletie therapie met stollingsfactorconcentraten heeft als belangrijkste bijwerking het ontstaan van remmende antistoffen tegen de desbetreffende stollingsfactor. In die situatie wordt de behandeling van de hemofilie ernstig bemoeilijkt. Voor behandeling van bloedingen in aanwezigheid van remmende antistoffen zijn zogenaamde ‘bypassing agents’ beschikbaar.
Nieuwe behandelmethoden voor hemofilie die niet gebaseerd zijn op toediening van de ontbrekende factor, zijn in ontwikkeling. Een recombinant gehumaniseerde, bispecifieke antistof, met zowel affiniteit voor factor IX als factor X, die de functie van factor VIII kan overnemen, is de eerste van deze nieuwe behandelingen die inmiddels wordt toegepast.
Al deze ontwikkelingen op het gebied van hemofilie hebben behandelkeuzes voor behandelaren uitgebreid.
Deze richtlijn is een document met aanbevelingen en instructies ter ondersteuning van de dagelijkse praktijk van de diagnostiek, behandeling en follow-up van hemofilie bij kinderen en volwassenen. Voorafgaande aan de ontwikkeling van de richtlijn zijn uitgangsvragen geformuleerd. Deze vragen volgen uit een knelpuntinventarisatie verricht bij medisch specialisten, verpleegkundig specialisten en paramedici werkzaam binnen het vakgebied en in de Hemofiliebehandelcentra als ook samen met patiënten (vertegenwoordigers). De meest relevante knelpunten zijn uitgewerkt tot uitgangsvragen. Dit betreft een beperkt aantal problemen in de dagelijkse praktijk rond het diagnostisch, therapeutisch en follow-up beleid van patiënten met hemofilie. De uitgangsvragen vormen de basis voor de verschillende modules van deze richtlijn.
De richtlijn beoogt derhalve niet een volledig leerboek te zijn. De richtlijn is bedoeld om aanbevelingen te geven, daar waar in de dagelijkse praktijk de belangrijkste knelpunten bestaan en tracht een betere uniformiteit van behandeling van deze patiënten in Nederland te bewerkstelligen, met name ook op die gebieden waar bewijs ontbreekt. Deze richtlijn is zoveel mogelijk gebaseerd op wetenschappelijke onderzoek of consensus. Het niveau van bewijsvoering staat vermeld in de tekst.
Deze richtlijn is bestemd voor alle professionals die betrokken zijn bij de diagnostiek, behandeling en begeleiding van patiënten met hemofilie, zoals internisten (hematologen en vasculair geneeskundigen), kinderartsen, hemofilie verpleegkundigen/verpleegkundig specialisten, klinisch genetici, gynaecologen, orthopeden, en fysiotherapeuten.
Voor het ontwikkelen van de richtlijn is in 2017 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met hemofilie en vertegenwoordigers van de patiënten (zie hiervoor de samenstelling van de werkgroep). De werkgroep leden zijn door hun (beroeps)verenigingen gemandateerd voor deelname. De werkgroep werkte gedurende 2,5 jaar aan de totstandkoming van de richtlijn.
De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn en bestond uit:
Met ondersteuning van:
De werkgroep leden hebben schriftelijk verklaard of ze in de laatste vijf jaar een (financieel ondersteunde) betrekking onderhielden met commerciële bedrijven, organisaties of instellingen die in verband staan met het onderwerp van de richtlijn. In onderstaande tabel wordt een overzicht gegeven met de belangen van bij de ontwikkeling van deze richtlijn betrokken personen.
|
Naam |
Belangen |
|
H.C.J. Eikenboom |
Onderzoekbeurs: CSL Behring Sprekersgeld: Roche, Celgene Reisbeurs: Roche |
|
E.A.M. Beckers |
Onderzoekbeurs: Bayer |
|
M.H. Cnossen |
Onderzoek-, onderwijs- en reisbeurzen: Pfizer, Baxter/Baxalta/Shire, Bayer Schering Pharma, CSL Behring, Sobi Biogen, Novo Nordisk, Novartis, Nordic Pharma Steering board: Roche, Bayer |
|
M. Coppens |
Investigator industrie studies: uniQure, Bayer, Roche Adviesraad: CSL Behring, Novo Nordisk, UniQure Steering committee: uniQure Sprekersgeld: Bayer, BMS-Pfizer Jurycommissie: SOBI |
|
C.J. Fijnvandraat |
Onderzoekbeurs: CSL Behring, NovoNordisk Adviesraad: Takedo, Novo Nordisk, Roche |
|
K. Fischer |
Onderzoekbeurs: Bayer, Pfizer, Baxter/Shire, NovoNordisk, Biogen Investigator industrie studies: Roche, Biogen/Sanofi, CSL Behring, Bayer Adviesraad: Bayer, Baxter, Biogen, CSL Behring, Freeline, NovoNordisk, Pfizer, Roche, SOBI Sprekersgeld: Bayer, Baxalta/Shire, SOBI/Biogen, CSL Behring, Octapharma, Pfizer, NovoNordisk |
|
K.P.M. van Galen |
Onderzoekbeurs: CSL Behring, Bayer |
|
F.C.J.I. Heubel-Moenen |
geen |
|
M.J.H.A. Kruip |
Onderzoekbeurs: Pfizer, Sobi, Boehringer Ingelheim, Daiichi Sankyo, Bayer |
|
B.A.P. Laros-van Gorkum |
Onderzoekbeurs: Baxter, CSL Behring |
|
F.W.G. Leebeek |
Onderzoekbeurs: CSL Behring, Takeda (Shire), uniQure Consultant: uniQure, BioMarin, Takeda Lid DSMB: Roche |
|
E.P. Mauser-Bunschoten |
Onderzoeksgeld: Baxter |
|
F.J.M. van der Meer |
Ondersteuning HemoNed register: CSL Behring, Pfizer, Bayer, NovoNordisk, Sobi, Roche, Octapharma, Sanquin |
|
K. Meijer |
Onderzoekbeurs: Bayer, Pfizer, Sanquin, Sprekersgeld: Bayer, Sanquin, Boehringer Ingelheim, BMS, Aspen Reisbeurs: Bayer Consultant: uniQure |
|
L. Nieuwenhuizen |
geen |
|
C.H. van Ommen |
geen |
|
M. Peters |
geen |
|
S.E.M. Schols |
geen |
|
R.E.G. Schutgens |
Onderzoekbeurs: Bayer, Takeda, NovoNordisk, CSL Behring, Pfizer Adviesraad: Bayer, Sobi, NovoNordisk |
|
L.F.D. van Vulpen |
Onderzoekbeurs: CSL Behring, NovoNordisk |
|
P.F. Ypma |
geen |
|
D.E. Fransen van de Putte |
geen |
|
M.F.C.M. Knapen |
geen |
|
H.M.J. van der Linden-van der Zwaag |
geen |
|
P. Kleijn |
geen |
|
M.A. Timmer |
geen |
|
A.G.M. Neuman-van Eijk |
geen |
|
N. Uitslager |
Onderzoekbeurs: Sobi Adviesraad: Sobi, CSL Behring Congrescommissie: Bayer |
|
S.L.A. Meijer |
geen |
|
A. Plat |
geen |
|
R.A.A. Mathôt |
Onderzoekbeurs: Baxter/Baxalta/Shire/Takeda, Bayer, CSL Behring, Sobi Consultant: Bayer, CSL Behring, Merck Sharp & Dohme, Baxter/Baxalta/Shire/Takeda, Zeria |
|
A. Tuinenburg |
geen |
Methode ontwikkeling:
Voor de uitgangsvragen is waar mogelijk een systematische literatuursearch verricht. Voor meerdere uitgangsvragen is afgezien van een systematische search vanwege het ontbreken van voldoende gepubliceerde studies of vanwege de beschikbaarheid van internationale evidence-based richtlijnen of systematische reviews. Dit is in de richtlijn per uitgangsvraag aangegeven.
Werkwijze:
Er werd voor afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in de elektronische database van Medline. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekactie of gebruikte trefwoorden van de zoekactie en de gehanteerde selectiecriteria zijn te vinden in de module van de desbetreffende uitgangsvraag.
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn is/wordt digitaal verspreid onder alle relevante beroepsgroepen. Daarnaast wordt er een toelichting op de richtlijn aangeboden aan het Nederlands Tijdschrift voor Hematologie. Ook is de richtlijn in te zien op de website van de Nederlandse Vereniging voor Hematologie en zal een link naar de richtlijn geplaatst worden op de website van de Nederlandse Vereniging voor Kindergeneeskunde
In deze richtlijn worden de patiënten vertegenwoordigd door afgevaardigden van de Nederlandse Vereniging van Hemofilie-Patiënten (NVHP).
Het grote aantal nieuwe stollingsconcentraten, de ontwikkelingen in de behandeling van hemofilie en de lange termijn sinds de vorige richtlijn, gepubliceerd in 2009, maakt de herziening van de richtlijn Diagnostiek en Behandeling van Hemofilie noodzakelijk. Beoogd is uniformiteit in behandeling te creëren met een richtlijn die door de verschillende specialismen en beroepsgroepen gedragen wordt.
De Nederlandse Vereniging van Hemofiliebehandelaars heeft daarom het initiatief genomen een multidisciplinaire, ‘evidence-based’ richtlijn te ontwikkelen voor het beleid bij hemofilie.
|
Diagnostiek |
||
|
1 |
In welke gevallen moet laboratoriumdiagnostiek worden ingezet naar hemofilie? |
|
|
2 |
Hoe wordt de diagnose hemofilie bevestigd? |
|
|
3 |
Welke vervolg laboratoriumonderzoeken moeten verricht worden bij patiënten met hemofilie? |
|
|
Behandeling |
||
|
4 |
Wanneer en hoe wordt profylactische behandeling gegeven bij hemofilie? |
|
|
5 |
Voor welke medicatie moet worden gekozen bij welke hemofilie patiënt zonder remmers? |
|
|
6 |
Hoe is de behandeling bij een bloeding bij hemofilie? |
Tabel 6.1 Tabel 6.2 |
|
7 |
Hoe is de behandeling rondom een ingreep of operatie? |
Tabel 7.1 Tabel 7.2 |
|
8 |
Hoe is de prenatale diagnostiek en het beleid in de zwangerschap en bij de partus van hemofilie draagsters? |
Tabel 8.1 |
|
9 |
Behandeling bij remmers |
|
|
9.1 |
Hoe kan de kans op vorming van een remmer zo laag mogelijk worden gemaakt? |
|
|
9.2 |
Welke behandeling kan worden ingezet om een remmer te doen verdwijnen? |
|
|
9.3 |
Hoe is de preventie en behandeling van bloedingen bij een patiënt met een remmer? |
|
|
10 |
Wat zijn de diagnostische en therapeutische opties bij pijn en beperkingen veroorzaakt door gewrichts- en spierbloedingen in het verleden bij patiënten met hemofilie? |
|
|
11 |
Hemofilie bij ouderen |
|
|
11.1 |
Wat is de rol van de hemofiliebehandelaar in het cardiovasculair risicomanagement? |
|
|
11.2 |
Welke beleid voor chronische hepatitis C wordt bij patiënten met hemofilie geadviseerd? |
|
|
11.3 |
Moeten hemofilie patiënten deelnemen aan het bevolkingsonderzoek darmkanker (RIVM)? |
|
|
11.4 |
Wat zijn contra-indicaties voor het gebruik van DDAVP bij de oudere patiënt met hemofilie? |
|
|
12 |
Antitrombotisch beleid bij hemofilie |
|
|
12.1 |
Onder welke omstandigheden kunnen antitrombotica veilig worden gegeven bij de behandeling van acuut coronair syndroom in patiënten met hemofilie? |
|
|
12.2 |
Onder welke omstandigheden kunnen trombocyten aggregatieremmers veilig worden gegeven in het kader van secundaire preventie van coronaire events? |
|
|
12.3 |
Wanneer is medicamenteuze preventie van veneuze trombose aangewezen? |
|
|
12.4 |
Onder welke omstandigheden kunnen anticoagulantia veilig worden gegeven bij de therapeutische behandeling van reeds opgetreden trombose in patiënten met hemofilie? |
|
|
12.5 |
Wanneer zijn anticoagulantia aangewezen ter voorkoming van herseninfarct bij boezemfibrilleren? |
|
In welke gevallen moet laboratoriumdiagnostiek worden ingezet naar hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Deze vraag beschrijft bij welke patiëntcategorieën onderzoek naar hemofilie ingezet dient te worden.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
Bij patiënten met een verhoogde bloedingsneiging is onderzoek naar hemofilie geïndiceerd (expert opinion/consensus) |
|
C |
Bij personen die op grond van de familieanamnese hemofilie zouden kunnen hebben, is stollingsonderzoek geïndiceerd. (expert opinion/consensus) |
|
C |
Bij ernstige of matig-ernstige hemofilie dient diagnostiek zo spoedig mogelijk na de geboorte plaats te vinden (expert opinion/consensus) |
|
C |
Bij een geïsoleerd verlengde APTT is onderzoek naar hemofilie geïndiceerd (expert opinion/consensus) |
|
C |
Bij potentiële draagsters is stollingsonderzoek en DNA-onderzoek geïndiceerd (Plug, 2006) |
Samenvatting literatuur
Resultaten
De artikelen van Rodeghiero en Elbatarny beschrijven hoe de ernst van bloedingssymptomen gebruikt kunnen worden voor een inschatting van de voorafkans op het hebben van hemofilie (Rodeghiero, 2010; Elbatarny, 2014). De studie van Plug beschrijft dat draagsters met een verlaagd factor VIII/IX een bloedingsneiging hebben die vergelijkbaar is met milde hemofilie A en dat iets meer dan de helft van draagsters een normaal factor VIII/IX heeft en dat stollingstesten dus onvoldoende zijn om draagsterschap uit te sluiten (Plug, 2006).
Referenties
Bewijskracht literatuur
Level 3, consistent.
Zoeken en selecteren
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van een eerder gepubliceerde evidence-based richtlijnen, aangevuld met bij de auteurs bekende relevante reviews.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Geen evidence tabellen, omdat er geen systematisch literatuuronderzoek werd gedaan.
Overwegingen
Ad aanbeveling 1
Bij patiënten met een verhoogde bloedingsneiging dient onderzoek naar hemofilie en andere stollingsziektes ingezet te worden. Indien er sprake is van een recent ontstane bloedingsneiging dient een verworven oorzaak van hemofilie overwogen te worden. Het is niet goed aan te geven welke mate van bloedingsneiging groot genoeg is om dit onderzoek in te zetten en dit zal daarom op individuele basis bepaald moeten worden. Het is te overwegen om de bloedingsneiging te kwantificeren met behulp van een bloedingsscore. De meest gebruikte bloedingsscore is de Bleeding Assessment Tool (BAT) van de International Society on Thrombosis and Haemostasis (ISTH; Rodeghiero, 2010). Bij een totale score van < 4 (volwassen mannen), < 6 (volwassen vrouwen) en < 3 (kinderen) is de kans op het hebben van hemofilie laag (Elbatarny, 2014). Toch is de ISTH-BAT nog onvoldoende gevalideerd om op basis van de bovengenoemde afkapwaardes af te zien van laboratoriumdiagnostiek naar hemofilie. Het belangrijkste probleem van de ISTH-BAT lijkt dat deze onvoldoende gevoelig is bij patiënten die in hun leven weinig blootgesteld zijn aan ingrepen waarbij een stollingsziekte manifest zou worden; dit geldt met name voor kinderen. Hoewel er voor kind-specifieke bloedingsscores ontwikkeld zijn, geldt ook voor deze scores dat ze onvoldoende gevalideerd zijn om op basis van een lage score af te zien van laboratoriumdiagnostiek (Bowman, 2009)
Ad aanbeveling 3
Indien er sprake is van een ernstige of matig ernstige vorm van hemofilie in de familie, dan dient dit onderzoek zo spoedig mogelijk na de geboorte ingezet te worden, tenzij door middel van prenatale diagnostiek al eerder is uitgesloten dat de jongen hemofilie heeft. Voor dit stollingsonderzoek kan navelstrengbloed gebruikt worden. Indien hiermee de diagnose hemofilie gesteld wordt, dan dient deze nadien bevestigd te worden met stollingsonderzoek via venapunctie om fout-positieve uitslagen te voorkomen. DNA-onderzoek is over het algemeen hiervoor niet noodzakelijk. Gezien de stijging van factor IX dient bij hemofilie B dient dit ook later (na ongeveer 6 maanden) nog eens herhaald te worden.
Ad aanbeveling 4
Een geïsoleerd verlengde geactiveerde partiële trombopastinetijd (APTT) kan berusten op een circulerend lupus anticoagulans, op een deficiëntie van stollingsfactor VIII, IX, XI of XII, van prekallikreïne of hoogmoleculair gewicht kininogeen (HMWK), of op een remmer/antistof tegen een van deze factoren. Bij een geïsoleerd verlengde APTT waar geen verklaring voor is, dient daarom hemofilie uitgesloten te worden, zelfs indien er bij de patiënt geen sprake is van een bloedingsneiging.
Ad aanbeveling 5
Hemofilie A en B zijn ziektes met een X-chromosoom gebonden overerving. Draagsters kunnen ook een verlaagd factor VIII of IX hebben dat gepaard kan gaan met een bloedingsneiging. Meestal betreft het een milde verlaging, maar er kan ook sprake zijn van een meer ernstige verlaging in het geval van asymmetrische lyonisatie. Iets meer dan de helft van de draagsters van hemofilie heeft een factor VIII of IX activiteit die binnen de normale referentiewaardes valt (Plug, 2006). Daarom dient de diagnose draagster van hemofilie gesteld te worden op grond van DNA-onderzoek.
Familieonderzoek dient bij voorkeur door de klinisch geneticus te worden verricht. DNA-onderzoek wordt in een familie bij voorkeur eerst gedaan bij een mannelijke hemofilie patiënt om de oorzakelijke mutatie te bepalen (Fransen van de Putte, 2015). Daarna kan er gericht DNA-onderzoek gedaan worden naar de familiaire mutatie bij (potentiele) draagsters. Verder dient bij mogelijke draagsters de factor VIII/IX activiteit bepaald te worden om het bloedingsrisico in te schatten, waarbij vrouwen met een factor VIII/IX < 50 IU/dL qua fenotype beschouwd worden als patiënten met hemofilie. De APTT als screeningstest is onvoldoende, omdat deze nog normaal kan zijn bij een mild verlaagde factor VIII/IX activiteit.
Ad aanbeveling 6
Het is gebruikelijk om DNA diagnostiek bij draagsters te verrichten boven de leeftijd van 16 jaar, conform richtlijnen klinische genetica
Hoe wordt de diagnose hemofilie bevestigd?
Aanbevelingen
Onderbouwing
Inleiding
De hier bedoelde diagnostiek is de diagnostiek die nodig is om de diagnose hemofilie A of B te stellen.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
Voor de diagnostiek van hemofilie A is factor VIII activiteitsbepaling nodig (expert opinion/consensus) |
|
C |
Voor de diagnostiek van hemofilie B is factor IX activiteitsbepaling nodig (expert opinion/consensus) |
|
C |
Factor VIII wordt bepaald met one-stage stoltest en chromogene bepaling; factor IX met one-stage stoltest (expert opinion/consensus) |
|
C |
DNA-diagnostiek is geïndiceerd bij hemofilie d.m.v. een techniek die ook mutaties detecteert die in gen promotor en intronen voorkomen (expert opinion/consensus) |
Samenvatting literatuur
Resultaten
De reviews van Peyvandi, Duncan en Rodgers beschrijven de verschillen tussen de ‘one-stage’ stoltest en de chromogene bepaling voor het meten van de factor VIII activiteit (Peyvandi, 2016; Duncan, 2017; Rodgers, 2017). Het belang van DNA diagnostiek wordt onderbouwd in de richtlijnen van de Clinical Molecular Genetics Society (Keeny, 2010; Mitchell, 2010).
Referenties
Bewijskracht literatuur
Level 3, consistent.
Zoeken en selecteren
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van een eerder gepubliceerde evidence-based richtlijnen, aangevuld met bij de auteurs bekende relevante reviews.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Geen evidence tabellen, omdat er geen systematisch literatuuronderzoek werd gedaan.
Overwegingen
Ad aanbevelingen 1 en 2
Patiënten met hemofilie A en B hebben meestal een verlengde geactiveerde partiële tromboplastinetijd (APTT), terwijl de protrombinetijd (PT) normaal is. Echter, bij een factor VIII of IX activiteit van 30 IE/dL of hoger kan de APTT nog een normale stoltijd geven. Daarmee is de APTT niet geschikt als enige screeningtest. De diagnose hemofilie A wordt gesteld aan de hand van de factor VIII activiteit en de diagnose hemofilie B aan de hand van de factor IX activiteit. Afhankelijk van de ernst van de hemofilie zal een factor activiteit worden gevonden van < 1 IE/dL (ernstige hemofilie), 1-5 IE/dL (matig ernstig) en > 5 IE/dL (mild). Dit impliceert dat een draagster met een factor VIII c.q. factor IX activiteit < 50 IE/dL beschouwd wordt als een patiënte met hemofilie.
Factor VIII kan ook verlaagd zijn bij de ziekte van von Willebrand (VWD) en derhalve dient VWD te zijn uitgesloten bij de diagnose hemofilie A. Bij VWD type 2N (Normandy) is de binding tussen de Von Willebrand Factor (VWF) en factor VIII gestoord. Bij deze ziekte is de factor VIII activiteit verlaagd en zijn de VWF-antigeen en -activiteit meestal normaal. Bij twijfel ten aanzien van de diagnose hemofilie A, op basis van (familie)anamnese of indien met DNA-onderzoek geen oorzakelijke factor VIII mutatie gevonden is, moet de ziekte van von Willebrand (type 2N) worden uitgesloten d.m.v. tenminste gericht stollingsonderzoek en zo mogelijk DNA-onderzoek.
Ad aanbeveling 3
De factor VIII activiteit kan worden gemeten in een ‘one-stage’ stoltest of met chromogene reagentia (Peyvandi, 2016; Duncan, 2017; Rodgers, 2017). Enerzijds is het wenselijk om gebruik te maken van een zo fysiologisch mogelijk systeem wat neerkomt op de ‘one-stage’ stoltest. Anderzijds is bekend dat in ongeveer één derde van de gevallen van milde hemofilie A de concentratie van factor VIII in de ‘one-stage’ stoltest overschat wordt ten opzichte van de chromogene test. In deze gevallen lijkt de chromogene test dus beter. Dit wordt onder meer veroorzaakt door de langere incubatietijd in de chromogene test, waardoor deze gevoeliger is voor instabielere vormen van factor VIII. Om deze reden wordt aanbevolen om bij patiënten met een matig ernstige of milde hemofilie A eenmalig tegelijk de factor VIII activiteit te bepalen met zowel de ‘one-stage’ stoltest als met de chromogene bepaling. Bij de bepaling van ernst van de hemofilie en de behandeling wordt uitgegaan van de laagste van de 2 bepalingen om onderbehandeling te voorkomen.
Ook de factor IX activiteit kan bepaald worden met zowel een ‘one-stage’ stoltest als een chromogene bepaling. De waarde van de chromogene bepaling in de diagnostiek van hemofilie B is op dit moment nog onvoldoende duidelijk, zodat deze op dit moment nog niet geadviseerd wordt.
Ad aanbeveling 4
DNA-diagnostiek naar de in de familie voorkomende oorzakelijke factor VIII of factor IX mutatie wordt geadviseerd en dient in ieder geval bij één patiënt in de familie te zijn verricht (Keeny, 2010; Mitchell, 2010), waarbij de voorkeur uitgaat naar een mannelijke patiënt boven een (obligate) draagster (Franse van de Putte, 2015). DNA-diagnostiek dient de worden verricht in een daartoe geaccrediteerd DNA laboratorium (zie www.dnadiagnostiek.nl). Het belangrijkste doel van DNA-diagnostiek is het opsporen van draagsters, opdat zij adequaat voorgelicht kunnen worden ten aanzien van de opties voor prenatale of pre-implantatie genetische diagnostiek en de adviezen voor de wijze en locatie van bevalling. Voorlichting over prenatale of preimplantatie genetische diagnostiek als ook het familieonderzoek dient door een klinisch geneticus te worden verricht. De oorzakelijke DNA mutatie bepaalt ten dele de kans op remmervorming en kan daarom voor prognostische redenen bepaald worden.
Welke vervolg laboratoriumonderzoeken moeten verricht worden bij patiënten met hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
De hier bedoelde diagnostiek is de diagnostiek die nodig is in de follow-up van patiënten die bekend zijn met de diagnose hemofilie.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
De ontwikkeling van remmende antistoffen dient vervolgd te worden (aanbevelingen 1,2,3,4) (Collins, 2013) |
|
C |
Remmende antistoffen worden gemeten met de Bethesda-assay met de Nijmegenmodificatie (Collins, 2013; Verbruggen, 1995) |
|
C |
Keuze tussen ‘one-stage‘ stoltest of chromogene bepaling bij substitutietherapie is afhankelijk van het gebruikte stollingsfactorconcentraat (Peyvandi, 2016) |
|
C |
Bij gebruik van veilige stollingsconcentraten is geen screenen op HIV en virale hepatitis nodig (expert opinion/consensus) |
Samenvatting literatuur
Resultaten
De richtlijn van de UK Haemophilia Centre Doctors Organization beschrijft de afwegingen ten aanzien van de frequentie van het testen op remmende antistoffen (Collins, 2013). De studie van Verbruggen et al. laat zien dat de Nijmegenmodificatie van de Bethesda-assay specifieker is dan de reguliere Bethesda-assay, met name in de lage range (Verbruggen, 1995). De review van Peyvandi et al. beschrijft de verschillen tussen de ‘one-stage’ stoltest en de chromogene test (Peyvandi, 2016). De Nederlandse richtlijn Hemofilie uit 2009 beschrijft de Nederlandse consensus met betrekking tot het screenen op HIV en virale hepatitiden (Schutgens, 2009).
Referenties
Bewijskracht literatuur
Level 3, consistent.
Zoeken en selecteren
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van een eerder gepubliceerde evidence-based richtlijn, aangevuld met bij de auteurs bekende relevante reviews.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Geen evidence tabellen, omdat er geen systematisch literatuuronderzoek werd gedaan.
Overwegingen
Ad aanbevelingen 1-4
De incidentie van remmers tegen exogeen factor VIII bij tevoren niet behandelde patiënten met ernstige hemofilie A varieert tussen 20-45% in verschillende cohorten. In grote onderzoeken blijkt de mediaan te liggen na 10-15 expositiedagen. Na 50-75 expositiedagen wordt een plateau bereikt, waarna het optreden van antistoffen erg zeldzaam is (Collins, 2013). De incidentie van remmers tegen factor IX is veel lager en deze remmers tegen factor IX zijn geassocieerd met allergische reacties en anafylaxie, vooral bij volledige gendeleties. Bij matig ernstige en milde hemofilie A is de incidentie van remmers lager: 13% na 100 expositiedagen (Eckhardt, 2013). De remmerontwikkeling bij deze groep patiënten kent geen plateaufase na 50-75 expositiedagen, maar lijkt vooral samen te hangen met de onderliggende mutatie en intensieve behandelperiodes zoals operaties (Eckhardt, 2013; Van Velzen, 2017). Tenslotte kan een plotseling toename in de ernst van het bloedingspatroon of een afname van de opbrengst en/of halfwaardetijd van het factorconcentraat een aanwijzing zijn voor de ontwikkeling van een remmer.
Ad aanbeveling 5
Voor de bepaling van de remmende antistoffen dient gebruikt gemaakt te worden van de Bethesda bepaling (Verbruggen, 1995). Met deze test worden alleen functionele/neutraliserende antistoffen aangetoond. Om de specificiteit, met name in de lage range, te verbeteren dient de Nijmegen modificatie gebruikt te worden.
Ad aanbeveling 6
Modificaties van het factor VIII of IX molecuul in recombinante stollingsfactorconcentraten kunnen leiden tot onjuiste bepaling van de factor VIII of IX plasmaconcentratie bij gebruik van de ‘one-stage’ stoltest. Dit probleem kan deels ondervangen worden door gebruik te maken van een chromogene factor assay. Dit kan per preparaat verschillen, maar ook afhankelijk zijn van het gebruikte reagens. Ook voor bepaling van de factor IX activiteit bestaat zowel een chromogene bepaling als een ‘one-stage’ stoltest. Het is op dit moment nog onvoldoende duidelijk voor welke factorconcentraten de chromogene bepaling gebruikt moet worden. Ook chromogene bepalingen zijn niet bij elk factorconcentraat betrouwbaar toepasbaar. De keuze van test dient per laboratorium te worden afgewogen en afgestemd op het gebruikte stollingsfactorconcentraat.
Ad aanbeveling 7
Voor de introductie van adequate virusinactivatie van stollingsproducten in 1992 waren stollingsfactorconcentraten besmet met het hepatitis-C-virus (HCV; Schutgens, 2009). Patiënten die zijn behandeld met stollingsfactorconcentraten vervaardigd uit grote plasmapools, zijn in vrijwel alle gevallen besmet met het HCV-virus. Van degenen die met cryoprecipitaat zijn behandeld, is dit percentage 66%. In de periode 1979 tot 1986 is ongeveer 16% van de Nederlandse hemofilie patiënten die werden behandeld met stollingsfactorconcentraten of cryoprecipitaat, besmet met het humaan immunodeficiëntievirus (HIV). Voor de introductie van antiretrovirale therapie in 1996 is het merendeel van hen overleden. Bij vrijwel alle patiënten die in het verleden in Nederland zijn behandeld met onveilige stollingsfactorconcentraten is bekend of zij hierbij geïnfecteerd geraakt zijn. Echter, bij patiënten die behandeld zijn in niet-westerse landen moet deze diagnostiek nog steeds plaatsvinden.
Wanneer en hoe wordt profylactische behandeling gegeven bij hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Bij hemofilie zijn bloedingen meestal gelokaliseerd in één van de zes grote gewrichten: ellebogen, knieën en enkels. Recidiverende gewrichtsbloedingen leiden uiteindelijk tot hemofilie-artropathie, die vaak ernstig en invaliderend is. De mate van gewrichtsschade wordt bepaald door de ernst van de hemofilie en door het aantal gewrichtsbloedingen. Profylactische toediening van stollingsfactorconcentraat heeft als doel het aantal bloedingen te verminderen, waardoor op termijn de gewrichtsschade beperkt kan worden.
Primaire profylaxe wordt gedefinieerd als langdurige profylaxe gestart na de eerste gewrichtsbloeding en/of voor de leeftijd van 3 jaar (Blanchette, 2014). Primaire profylaxe wordt meestal vooral gestart bij patiënten met ernstige hemofilie, omdat bij hen het risico op spontane bloedingen groot is, maar soms ook bij matig-ernstige of milde hemofilie.
Secundaire profylaxe is langdurige profylaxe gestart na de leeftijd van 3 jaar of na 2 of meer gewrichtsbloedingen (Blanchette, 2014). Secundaire profylaxe kan gestart worden bij alle hemofilie patiënten die frequent gewrichtsbloedingen of ernstige weke delen bloedingen hebben, bij recidiverende bloedingen in 1 gewricht, bij chronische synovitis, of bij intensieve fysiotherapie of revalidatie. Tertiaire profylaxe wordt gestart bij volwassenen met hemofilie om progressie van artropathie door herhaalde gewrichtsbloedingen te voorkomen. Het onderscheid tussen secundaire en tertiaire profylaxe is niet altijd even duidelijk in de literatuur.
Conclusies
|
SORT Grade |
Conclusie |
|
A |
Het starten van primaire profylactische behandeling met factor VIII-concentraat op jonge leeftijd resulteert in minder gewrichtsbloedingen, minder artropathie en verbeterde kwaliteit van leven (Manco-Johnson, 2007; Gringeri, 2011) |
|
C |
Er is geen verschil in bloedingsfenotype op jonge leeftijd tussen kinderen met ernstige/matig ernstige hemofilie A en hemofilie B (Clausen, 2014) |
|
C |
Profylactische behandeling onder de leeftijd van 3 jaar of voor de tweede of derde gewrichtsbloeding zorgt voor betere gewrichts-uitkomsten bij lichamelijk onderzoek (Gilbert score) en bij radiologisch onderzoek (Petterson score) (Kreuz, 1998; Astermark, 1999; Gringeri, 2011; Fischer, 2002; Yee, 2002) |
|
C |
Bij ernstige hemofilie patiënten uit Zweden met hoge dosis-profylaxe is het verbruik van SHL factorconcentraat hoger (mediaan 4000 E/kg/jaar versus 2100 E/kg/jaar), en het mediaan aantal bloedingen (2,5 versus 10 per 5 jaar) en de HJHS score (4 versus 9 van 144 punten) lager dan bij Nederlandse patiënten met intermediaire dosis-profylaxe, terwijl de kwaliteit van leven gelijk is (Fischer, 2013) |
|
C |
Starten met profylaxe met 1 tot 2 infusies per week, waarna binnen 3 tot 6 maanden wordt opgebouwd naar het volledige schema of een extra infusie wordt toegevoegd na elke bloeding leidt tot minder frequente plaatsing van port-a-caths (PACs) dan direct starten met een volledig profylaxe schema van ≥ 3 infusies per week bij hemofilie A en ≥ 2 infusies per week bij hemofilie B patiënten. (34% en 22%, respectievelijk versus 88%) (Nijdam, 2015) |
|
C |
Starten met profylaxe 1x per week en opbouwen bij een volgende gewrichts- en of andere belangrijke bloeding zorgt op de leeftijd van 4 jaar voor 2 gewrichtsbloedingen meer dan direct volledige primaire profylaxe en starten met profylaxe 1x per week en zo spoedig mogelijk opbouwen in 3 tot 6 maanden (Nijdam, 2015) |
|
C |
On demand behandeling bij volwassenen met ernstige hemofilie gaat gepaard met meer bloedingen per jaar dan profylaxe behandeling (27,9 versus 0) (Manco-Johnson, 2013) |
|
C |
On demand behandeling bij volwassenen met ernstige hemofilie gaat gepaard met meer progressie van artropathie bij lichamelijk en radiologisch onderzoek dan profylaxe behandeling. (HJHS: 23 versus 14 en Pettersson: 16 versus 5, respectievelijk) (Nijdam, 2016) |
|
C |
Secundaire en tertiaire profylaxe vermindert het aantal bloedingen en de pijn, en verbetert de functionaliteit van de gewrichten en de kwaliteit van leven bij patiënten met ernstige en matig ernstige hemofilie (Nugent, 2018) |
Samenvatting literatuur
Resultaten
Primaire profylaxe bij kinderen
Twee prospectieve gerandomiseerde studies bij ernstige hemofilie A patiënten tonen aan dat het starten van profylactische behandeling met factor VIII-concentraat op jonge leeftijd resulteert in minder gewrichtsbloedingen, minder artropathie en verbeterde kwaliteit van leven (Gringeri, 2011; Manco-Johnson, 2007). Voor kinderen met ernstige hemofilie B zijn er geen vergelijkbare studies voorhanden door het kleine aantal patiënten. Een studie van Clausen et al. liet echter geen verschil in bloedingsfenotype zien op jonge leeftijd tussen kinderen met ernstige en matig ernstige hemofilie A en hemofilie B (Clausen, 2014).
Vijf studies bestudeerden de effecten van de start leeftijd van profylaxe op de gewrichten met behulp van lichamelijk onderzoek (Gilbert score) en/ of radiologisch onderzoek (Petterson score) (Fischer, 2016). Drie studies rapporteerden een betere uitkomst bij jongens die profylaxe voor de leeftijd van 3 jaar startten (Gringeri, 2011; Astermark, 1999; Kreuz, 1998); de twee andere studies beschreven een betere uitkomst bij jongens die profylaxe voor de tweede of derde gewrichtsbloeding startten (Fischer, 2002; Yee, 2002).
Er zijn geen prospectieve (gerandomiseerde) interventie studies die de effectiviteit van de verschillende profylactische strategieën bestuderen. Drie observationele studies zijn relevant:
In een prospectieve studie van Collins et al. bij patiënten met profylaxe, nam het risico op een bloeding toe bij de patiënten indien het factor VIII plasma gehalte gedurende langere tijd < 1 E/dL was (Collins, 2009).
In de retrospectieve studie van Fischer et al. werden de uitkomsten en kosten vergeleken tussen het Nederlandse intermediaire- en het Zweedse hoge-dosis profylaxe regime bij patiënten met ernstige hemofilie A of B, die geboren waren tussen 1970 en 1994 (Fischer, 2013). Er werden 78 Nederlandse en 50 Zweedse patiënten geïncludeerd (mediane leeftijd 24 jaar, range 14-37 jaar). De Nederlandse patiënten gebruikten minder stollingsfactorconcentraat (mediaan 2100 E/kg/jaar versus 4000 E/kg/jaar) in de laatste 5 jaar. Het mediaan aantal bloedingen en de mediane HJHS score was respectievelijk 10 per 5 jaar en 9,0 punten van 144 bij de Nederlandse patiënten, en 2,5 per 5 jaar en 4,0 van 144 punten bij de Zweedse patiënten. De kwaliteit van leven was in beide cohorten gelijk. De Zweedse patiënten begonnen eerder met profylaxe (mediaan 1,5 jaar versus 4,5 jaar).
In de retrospectieve studie van Nijdam et al. werden 3 verschillende strategieën onderscheiden t.a.v. de frequentie van infusies bij het starten van profylaxe met standaard stollingsfactorconcentraat: 1. direct starten met een volledig profylaxe schema van ≥ 3 infusies per week bij hemofilie A en ≥ 2 infusies per week bij hemofilie B patiënten, 2. starten met 1 tot 2 infusies per week, waarna binnen 3 tot 6 maanden wordt opgebouwd naar het volledige schema, en 3. starten met 1 tot 2 infusies per week, waarbij een extra infusie wordt toegevoegd na elke bloeding. Het aantal benodigde Port-a-caths nam af per strategie (88% vs 34% vs 22%) (Nijdam, 2015). Kinderen met profylaxe volgens strategie 3 hadden t.o.v. strategie 1 en 2, op de leeftijd van 4 jaar minder infusies gekregen, maar wel 2 extra gewrichtsbloedingen.
In een recente studie van Buckley hadden kinderen met hemofilie in Amerika met een PAC vaker infecties (29% versus 17%), en trombose (6% versus 1%) en kwamen zij vaker in het ziekenhuis dan kinderen met hemofilie zonder PAC (Buckley, 2018).
Continuering van profylaxe bij volwassenen
Nijdam et al. onderzocht de gewrichtsstatus van patiënten met ernstige hemofilie 10 jaar na het stoppen van profylaxe en vergeleek die met de gewricht status van patiënten die wel doorgingen met profylaxe. Alhoewel het zelf gerapporteerde aantal bloedingen en functionele beperkingen in beide groepen gelijk waren, hadden on demand patiënten duidelijk meer progressie van artropathie bij lichamelijk en radiologisch onderzoek dan patiënten met profylaxe (HJHS: 23 vs. 14 en Pettersson: 16 vs 5, respectievelijk) (Nijdam, 2016).
Secundaire en tertiaire profylaxe
Er zijn enkele prospectieve gerandomiseerde studies die het effect van secundaire en tertiaire profylaxe hebben onderzocht op onder andere het aantal bloedingen, functionaliteit van de gewrichten, pijn, activiteit, en de kwaliteit van leven bij patiënten met ernstige hemofilie. Alle studies laten zien dat secundaire of tertiaire profylaxe een gunstig effect hebben op alle bovengenoemde punten (Nugent, 2018). Een van de studies is de SPINART studie. Dit was een prospectieve gerandomiseerde studie waarbij on demand therapie werd vergeleken met secundaire profylaxe bij 84 adolescenten en volwassenen (gemiddelde leeftijd 30,6 jaar) met ernstige hemofilie A (mediane behandelperiode: 1,7 jaar), die in de afgelopen 5 jaar > 12 maanden geen profylaxe hadden gebruikt. Met profylaxe was het mediaan aantal bloedingen en het mediaan aantal bloedingen per jaar significant lager dan met on demand therapie, respectievelijk 0 versus 54,5 en 0 versus 27,9 (Manco-Honson, 2013). Verder hadden de profylaxe patiënten minder pijn, betere functionaliteit van de 6 index gewrichten (CAJAS score), waren meer actief en tevreden. Er was echter geen verschil tussen beide groepen in vermindering van progressie van gewrichtsschade afgebeeld met MRI (Manco-Johnson, 2017).
Referenties
Bewijskracht literatuur
Level 1 en Level 3
Zoeken en selecteren
Er werd voor deze uitgangsvraag geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van gepubliceerde richtlijnen (Fischer, 2016; Nordic Haemophilia Guideline 2015; Richards, 2010), aangevuld met bij de auteurs bekende relevante studies.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Geen evidence tabellen, omdat er geen systematisch literatuuronderzoek werd verricht.
Overwegingen
Primaire profylaxe bij kinderen
Alleen bij kinderen met ernstige hemofilie A is bewezen dat het starten van profylactische behandeling met factor VIII-concentraat op jonge leeftijd resulteert in minder gewrichtsbloedingen, minder artropathie en verbeterde kwaliteit van leven (Gringeri, 2011; Manco-Johnson, 2007). Aangezien de studie van Clausen et al. geen verschil in bloedingsfenotype liet zien op jonge leeftijd tussen kinderen met ernstige/ matig ernstige hemofilie A en hemofilie B, is ook bij kinderen met ernstige hemofilie B een vroege start van profylactische behandeling zinvol (Clausen, 2014).
Er is echter grote variatie in het tijdstip van de eerste gewrichtsbloeding: de mediane leeftijd is 1,7 jaar, met een spreiding van 0,2 tot 5,8 jaar (van Dijk, 2005). Een optimale start-strategie zal onderbehandeling voorkómen bij patiënten met een ernstig bloedingsfenotype en overbehandeling bij patiënten met een milder fenotype.
Het advies ten aanzien van de startleeftijd van primaire profylaxe varieert in de literatuur. Studies naar de effecten van startleeftijd op de gewrichten met behulp in van lichamelijk onderzoek (Gilbert score) en/ of radiologisch onderzoek (Pettersson score) laten betere uitkomsten zien bij jongens die profylaxe voor de leeftijd van 3 jaar startten of bij jongens die profylaxe voor de tweede of derde gewrichtsbloeding startten (Fischer, 2002; Yee, 2002). De factor VIII/IX SSC van de ISTH adviseert daarom primaire profylaxe te starten niet later dan onmiddellijk of kort na de eerste gewrichts- of spierbloeding en altijd onmiddellijk na de initiële behandeling van een hersenbloeding (Fischer, 2016).
Om het prikken bij kinderen te vergemakkelijken wordt vaak een PAC ingebracht. Een PAC heeft echter ook nadelen. Kinderen met een PAC hebben vaker een infectie en trombose dan kinderen zonder PAC. Ook bezoeken zij daardoor het ziekenhuis vaker (Buckley, 2018). Om het inbrengen van een PAC bij jonge kinderen te vermijden wordt regelmatig gestart met stollingsfactorconcentraat infusie 1x per week. De studie van Nijdam et al liet zien dat het ophogen van dosis frequentie alleen bij een gewrichts-, spier of andere belangrijke bloeding, zorgde voor 2 extra gewrichtsbloedingen op de leeftijd van 4 jaar (Nijdam, 2015). De gevolgen hiervan op de latere gewrichtsstatus is niet bekend. Indien de profylaxe frequentie binnen 3 tot 6 maanden wordt opgehoogd naar volledig, is het aantal gewrichtsbloedingen op de leeftijd van 4 jaar niet toegenomen, terwijl er minder PACs werden ingebracht (34%) t.o.v. het meteen volledig profylaxeschema (88%). De factor VIII/IX SSC van de ISTH adviseert daarom profylaxe met stollingsfactorconcentraat te starten met minimaal 1 of meerdere infusies per week. Indien gestart wordt met 1 infusie per week, moet de frequentie zo snel mogelijk opgehoogd worden, maar in ieder geval na elke gewrichts-, spier- of andere belangrijke bloeding (Fischer, 2016).
De startdosis van de profylaxe is afhankelijk van de beschikbare afvuleenheid. Aangezien de meeste kinderen jonger dan 3 jaar zijn bij start profylaxe en waarschijnlijk tussen de 10 en 15 kg wegen, is starten met 25 E/kg per infusie voldoende bij hemofilie A patiënten (overeenkomend 250 E) en 50 E /kg per infusie voor hemofilie B patiënten (overeenkomend met 500 E). Bij hemofilie A patiënten geeft 500 E 1 half-life (6-8 uur) extra bescherming. Aangezien bij hemofilie A patiënten een hoge dosis mogelijk geassocieerd is met de ontwikkeling van remmers wordt steeds vaker gestart met 250 E (Gouw, 2007; Iorio, 2010).
De uiteindelijke dosis en dosisinterval van profylactische infusies moet individueel bepaald worden en is afhankelijk van het doel van de behandeling, de kosten van de behandeling, het bloedingsfenotype, de dagelijkse activiteiten, veneuze toegang, en de beschikbare afvuleenheid van de stollingsfactorconcentraten. Alhoewel Collins et al. in een prospectieve studie lieten zien dat bij patiënten met profylaxe het risico op een doorbraakbloeding toenam indien het factor VIII plasma gehalte langer < 1 E/dL was, betekent dit vooralsnog niet automatisch dat bij alle patiënten de plasma factor VIII-concentratie altijd > 1 E/dL moet zijn (Collins, 2009). Er zijn grote interindividuele verschillen in het aantal en de ernst van gewrichtsbloedingen die nodig zijn om synovitis en vervolgens artropathie te ontwikkelen. Sommige patiënten met ernstige artropathie op MRI hebben nooit een klinische gewrichtsbloeding doorgemaakt, terwijl andere patiënten na zeer ernstige gewrichtsbloedingen geen artropathie ontwikkelen (Manco-Johnson, 2007). In de studie van Fischer et al. over de uitkomsten bij een hoge dosis-profylaxe en intermediaire dosis profylaxe waren de verschillen in de gewrichtsstatus na 5 jaar klein: de mediane HJHS score was 9,0 punten van 144 bij de Nederlandse patiënten, 4,0 van 144 punten bij de Zweedse patiënten. Echter, in het Zweedse model begonnen de kinderen op jongere leeftijd met profylaxe. De lange termijn effecten van de gewrichtsbloedingen zijn niet bekend (Fischer, 2013).
De meeste richtlijnen adviseren om kinderen met ernstige hemofilie A minimaal 3x per week of om de dag SHL factorconcentraat infusies toe te dienen en kinderen met ernstige hemofilie B minimaal 2x per week of om de 3 dagen (Fischer, 2016; Nordic Haemophilia Guideline 2015; Richards, 2010). De UKHCDO richtlijn adviseert om tenminste de dosis van SHL factorconcentraat te gebruiken die spontane bloedingen voorkomt. Als een 3x per week schema wordt gebruikt, wordt een hogere dosis op de derde dag niet geadviseerd. Het is effectiever om een extra infusie te geven, of over te gaan naar een om de dag infusie schema. Deze richtlijn adviseert verder om eventueel zeer actieve jongens of jongens met een minder frequent profylaxe schema en doorbraakbloedingen te behandelen met dagelijkse infusies met lage dosis stollingsfactorconcentraat om het factor VIII of IX gehalte > 1 E/dl te houden. Farmacokinetische metingen kunnen helpen om de dosis en dosisinterval te bepalen en te monitoren.
De profylaxe dagen zijn mede afhankelijk van de (sport) activiteiten van het kind. Bij evenementen als sportdag of schoolreisje kan het schema worden aangepast of wordt een extra gift toegediend. De profylaxe wordt bij voorkeur in de ochtenduren (dus vóór schooltijd) toegediend.
Continuering van profylaxe bij volwassenen
Er zijn wisselende opinies over de continuering van profylaxe bij volwassenen met ernstige hemofilie, met name bij volwassenen met een mild bloedingsfenotype.
Enkele prospectieve studies laten zien dat het continueren van profylaxe op de volwassen leeftijd het aantal bloedingen vermindert alsmede de progressie van artropathie (Nijdam, 2016; Manco-Johnson, 2013). Het wordt dan ook geadviseerd om indien mogelijk, de profylaxe te continueren na de adolescentie. Eventueel kan in sommige patiënten met een mild bloedingsfenotype, profylaxe intermitterend worden gegeven bij sport activiteiten en potentiele traumatische gebeurtenissen. Bij het optreden van spontane bloedingen moet de profylaxe echter weer herstart worden om het risico op artropathie te verkleinen.
Secundaire en tertiaire profylaxe
Secundaire en tertiaire profylaxe wordt toegepast bij ernstige, matig-ernstige en milde hemofilie, kan kortdurend of langdurig zijn en wordt eventueel gestart bij frequente gewrichtsbloedingen of ernstige weke delen bloedingen, bij recidiverende bloedingen in 1 gewricht, bij chronische synovitis, of bij intensieve revalidatie met fysiotherapie. Secundaire profylaxe vermindert het aantal bloedingen waardoor de mobiliteit en de pijn verbetert, het ziekteverzuim vermindert en de kwaliteit van leven toeneemt. De benodigde dosisbehoefte voor profylaxe bij volwassenen kan enorm variëren door de wisselende half-waarde tijd (8 tot 23 uur) (Richards, 2010). De dosis en dosisinterval is gebaseerd op het bloedingsfenotype en indien mogelijk op de individuele farmacokinetiek. Ook bij volwassenen adviseert de UKHCDO om de minimale hoeveelheid factorconcentraat te gebruiken die nodig is om bloedingen te voorkomen. Langdurige secundaire profylaxe is geïndiceerd na een intracraniële bloeding als de onderliggende aandoening niet gecorrigeerd kan worden.
Voor welke medicatie moet worden gekozen bij welke hemofilie patiënt zonder remmers?
Aanbevelingen
Onderbouwing
Inleiding
De medicamenten die op dit moment in Nederland ter beschikking zijn voor de behandeling van hemofilie, zijn onder te verdelen in de volgende groepen:
De keuze voor een bepaald product wordt bepaald door veiligheid, effectiviteit, beschikbaarheid en kosten van het product, evenals medische factoren zoals: veneuze toegang, halfwaardetijd product in patiënt, en psychologische factoren. Het laatste decennium is het aantal beschikbare producten sterk toegenomen en in de komende jaren zal dit met zekerheid nog verder worden uitgebreid.
In dit hoofdstuk gaan wij in op de standaard-halfwaarde en de langerwerkende stollingsfactorconcentraten en emicizumab.
Conclusies
|
SORT Grade |
Conclusie |
|
A |
Het risico op remmer vorming in previously untreated patients (PUPs) met hemofilie A is hoger bij tweede generatie dan bij derde generatie recombinante producten (Gouw, 2013; Volkers, 2019) |
|
B |
De kans op vorming van remmende antistoffen bij PUPs is mogelijk lager bij uit plasma bereid factor VIII dan tweede generatie recombinant factor VIII (Peyvandi, 2016) |
|
A |
De halfwaardetijd van recombinante producten wordt verlengd door PEGylering of koppeling aan lichaamseigen eiwitten (Lambert, 2018) |
|
n.v.t. |
De lange termijnseffecten van chronische en frequente blootstelling aan PEG zijn onbekend. |
|
A/B |
De kans op remmer ontwikkeling na switch naar een langerwerkend product is laag voor previously treated patients (Collins, 2016; Young, 2016) |
|
A |
In patiënten met hemofilie A zonder remmers is emicizumab even effectief als profylaxe met factor VIII in het voorkómen van bloedingen (Mahlangu, 2018) |
|
A |
In patiënten met matig-ernstige of milde hemofilie A zonder factor VIII remmers die een indicatie hebben voor profylactische behandeling is de behandeling met emicizumab veilig en effectief (Hermans, 2022) |
Samenvatting literatuur
Er zijn geen head-to-head vergelijkende studies tussen de verschillende type producten. Hieronder wordt een beschrijving gegeven van de beschikbare producten en hun kenmerken.
Resultaten
De meest gebruikte stollingsfactorconcentraten zijn de standaard-halfwaardetijd producten. Dit zijn factor VIII/IX producten met een halfwaardetijd 8 tot 15 uur voor factor VIII en 18 tot 24 uur voor factor IX. Plasma producten zijn veilig wat betreft HIV sinds 1985 en voor hepatitis C sinds 1992 (Mannucci, 1993).
Sinds de jaren ’90 zijn in Nederland stollingsfactoren beschikbaar die volgens recombinant-DNA-technieken worden gemaakt. De nieuwste generatie recombinante producten wordt geproduceerd zonder gebruik te maken van menselijke of dierlijke eiwitten, waardoor de kans op mogelijke overdracht van infectieuze agentia, zoals virussen en prionen, wordt geminimaliseerd. Uit plasma bereide stollingsfactorproducten hebben één of meer virus-inactiverende behandelingen ondergaan, waarbij virussen met een lipidenenvelop, zoals hepatitis C en HIV, worden geïnactiveerd (Lassila, 2016).
Tegenwoordig is het ontwikkelen van remmende antistoffen tegen de stollingsfactor de belangrijkste en meest uitdagende complicatie van behandeling met stollingsfactoren. Er zijn verschillende factoren die een rol spelen in de kans op remmer vorming. Of het type stollingsfactorproduct hier ook mee geassocieerd is blijft aanhoudend onderwerp van discussie. Observationele studies verschillen qua uitkomst door verschillen in studie opzet, studie periode, frequentie van remmer bepalingen en follow-up. In een systematische review uit 2010 naar de remmer ontwikkeling onder 420 jonge, niet eerder behandelde hemofilie A patiënten (previously untreated patients, PUPs) uit 24 studies toonde geen statistisch significante relatie met het type stollingsconcentraat (Iorio, 2010). De gepoolde incidentie voor remmer vorming in deze studie was 14% voor plasmaproducten en 27% voor recombinant factor VIII en bleek in de multivariate analyse vooral verklaard door verschillen in studieopzet. Deze relatie werd ook niet gevonden in twee grote observationele studies van na deze datum. De cumulatieve remmer ontwikkeling in respectievelijk 574 en 417 PUPs met hemofilie A in deze studies was 32% en 26% (Fischer, 2015; Gouw, 2013). In de studie van Gouw et al, de adjusted hazard ratio (HR) voor remmer ontwikkeling bij plasmaproducten in vergelijking met recombinante producten was 0,96 (95% betrouwbaarheidsinterval (BI) 0,62-1,49).
In de periode van 2010 tot 2014 is een gerandomiseerd onderzoek uitgevoerd naar het ontstaan van remmers bij plasma producten versus recombinante producten bij PUPs met hemofilie A, de SIPPET trial (Peyvandi, 2016). De cumulatieve incidentie van remmer vorming was 28,6% (95% BI 18,4-35,2%) voor behandeling met plasmaproducten en 44,5% (95% BI 19,6-37,2%) voor recombinant factor VIII. Deze data suggereren een hogere remmer vorming bij het gebruik van recombinante stollingsfactoren in PUPs. Hierbij dient opgemerkt te worden dat de remmer vorming in deze studie hoger is dan verwacht, mogelijk dat eerdere behandeling met bloedproducten hier een rol in speelt. In de SIPPET studie zijn alleen eerste (in Nederland niet meer verkrijgbaar) en tweede generatie (tijdens het opkweken van de cellijn nog gebruik van plasmacomponenten van menselijke afkomst) recombinante factor VIII producten gebruikt. Inmiddels zijn er ook veel derde generatie factoren op de markt, waarbij in het gehele productieproces geen gebruik meer wordt gemaakt van plasma componenten van menselijke afkomst. Een gecombineerde analyse van drie grote observationele studies onder 1109 PUPs laat zien dat het risico op remmer vorming bij Advate® (derde generatie recombinante factor VIII) lager is t.o.v. Kogenate®/Helixate® (tweede generatie producten) (adjusted HR 0,68; 95% BI 0,52-0,91) (Volkers, 2019). Ook in de eerder genoemde cohortstudie van Gouw et al. was het risico op remmer vorming hoger bij tweede generatie factoren t.o.v. derde generatie factoren (adjusted HR 1,60; 95% BI 1,08-2,37 en voor hoge-titer remmer vorming adjusted HR 1,79; 95% BI 1,09-2,94) (Gouw, 2013).
Tot slot is het van belang te realiseren dat de behandelstrategie in de SIPPET trial verschilt van de richtlijnen in Nederland. Patiënten werden later (mediaan 20 maanden) en minder intensief (slechts 15% op profylaxe en 7 exposities/jaar in de on demand patiënten) behandeld. De intensiteit van de behandeling is geassocieerd met het risico op remmer vorming.
Door modificatie van recombinante stollingsfactoren is het mogelijk de halfwaardetijd te verlengen en daarmee het dosisinterval te verlengen. Voor factor VIII resulteert dit in een gemiddeld 1,5-1,8 maal verlenging van de halfwaardetijd, en 3-5 maal voor factor IX (Lambert, 2018).
Het is van belang te realiseren dat de modificatie effect kan hebben op de stollingsfactorbepalingen en er grote discrepanties kunnen zijn tussen de one-stage assay en chromogene bepalingen.
Het chemisch koppelen van polyethyleenglycol (PEG) aan een doeleiwit is een veelgebruikte methode om de halfwaardetijd te verlengen. Er zijn drie gePEGyleerde factor VIII producten beschikbaar, BAX855 (Adynovate®, op basis van Advate®), BAY94-9027 (op basis van een B-domein-deleted rFVIII) en N8-GP (Esperoct®, op basis van B-domein-truncated rFVIII) met een halfwaardetijd van 14u, 19u en 18-19u respectievelijk (Coyle, 2014; Giangrande, 2017; Konkle, 2015). Er is één factor gePEGyleerd IX product, N9-GP (Refixia®), waarvan de gemiddelde halfwaardetijd 96u bedraagt (Tiede, 2017).
Het gebruik van PEG polymeren kan leiden tot de vorming van anti-PEG antilichamen, wat bij andere producten zoals PEG-asparaginase en PEG-uricase leidt tot een sterke verkorting van de halfwaardetijd van deze producten (Ganson, 2006, Garay, 2012). Door gebruik van PEG polymeren in voedsel en cosmetica, kan het zelfs voorkomen dat patiënten al antistoffen hebben vóór blootstelling aan het langwerkende product (Armstrong, 2007). In de huidige studies met gePEGyleerd factor VIII en factor IX wordt wel een hoger percentage antilichamen gevonden, dit zijn niet-neutraliserende antilichamen veelal gericht tegen PEG (Collins, 2014; Reding, 2017).
PEG is een lichaamsvreemde stof die niet biologisch afbreekbaar is. Het is onbekend of chronische en frequente blootstelling aan PEG zoals in de langerwerkende stollingsconcentraten aanwezig is, bij profylaxe in hemofilie patiënten, kan leiden tot negatieve effecten (Peyvandi, 2019; Verhoef, 2013). Lange termijn surveillance is nodig om hier meer inzicht in te krijgen. De huidige informatie van andere geregistreerde gePEGyleerde producten kan hier onvoldoende antwoord op geven, aangezien deze producten niet levenslang gebruikt worden.
Een ander theoretisch bezwaar van gePEGyleerde producten is het optreden van vacuolisatie in bepaalde celtypes waaronder epitheelcellen en de plexus choreoïdus (Agency, 2012). Dit is in toxicologische dierstudies van de huidige gePEGyleerde stollingsfactorproducten niet waargenomen (Lambert, 2018).
Factor VIII en IX kunnen ook gekoppeld worden aan lichaamseigen eiwitten om de halfwaardetijd te verlengen. De halfwaardetijd van zowel humaan IgG als albumine is ongeveer 3 weken door recycling via de neonatale Fc receptor. rFVIIIFc (Elocta®) is een recombinant factor VIII zonder het B-domein, covalent gekoppeld aan het Fc-domein van humaan immunoglobuline G1 met een gemiddelde halfwaardetijd van 19u (Mahlangu, 2014; Powell, 2012). rFIXFc (Alprolix®) is een fusie-eiwit van recombinant factor IX en het Fc-domein met een gemiddelde halfwaardetijd van 82u (Powell, 2013).
Het enige albumine-fusie eiwit dat beschikbaar is, is Idelvion® waarbij recombinant factor IX covalent gebonden is aan recombinant humaan albumine. Dit product heeft een gemiddelde halfwaardetijd van 102u (Santagostino, 2016).
Op basis van de huidige data lijkt de kans op het ontwikkelen van een remmer na switch naar een langerwerkend product voor previously treated patients (PTPs) laag. Er zijn geen data voor patiënten met een voorgeschiedenis van remmer ontwikkeling of bij een positieve familieanamnese voor remmer ontwikkeling (Collins, 2016).
Product typen/tabellen (Lambert, 2018; Peyvandi, 2019)
Hemofilie A
|
Molecuul naam |
Merknaam (stofnaam) |
Modificatie |
Gemiddelde t1/2 |
|
rFVIIIFc |
Elocta® (Efmoroctocog alfa) |
Fc-fusie |
19u |
|
BAX855 |
Adynovi® (Rurioctocog alfa Pegol) |
PEGylering |
14-16u |
|
N8-GP |
Esperoct® (Turoctocog Alfa Pegol) |
GlycoPEGylering |
18-19u |
|
BAY94-9027 |
Jivi® (Damoctocog alfa Pegol) |
PEGylering |
19u |
Hemofilie B
|
Molecuul naam |
Merknaam (stofnaam) |
Modificatie |
Gemiddelde t1/2 |
|
rFIXFc |
Alprolix® (Eftrenonacog alfa) |
Fc-fusie |
82u |
|
CSL654 (rIX-FP) |
Idelvion® (Albutrepenonacog alfa) |
Albumine fusie |
102u |
|
N9-GP |
Refixia® (Nonacog beta Pegol) |
GlycoPEGylering |
93u |
Hoewel nog steeds in de praktijk wordt gedoseerd o.b.v. lichaamsgewicht, worden ruwe schattingen van PK van een stollingsfactorconcentraat in een specifiek individu al langer toegepast. Verschillende studies en publicaties zijn verschenen t.a.v. het toenemend toepassen van farmacokinetisch (PK)-gestuurd doseren o.b.v. populatie PK modellen van stollingsfactorconcentraten door middel van Bayesiaanse analyse (NONMEM®) o.a. twee recente SSC aanbevelingsartikelen (Iorio, 2018, Ragni, 2018). Voordelen zijn meer geïndividualiseerd doseren, slechts “limited sampling” noodzakelijk om individuele PK parameters vast te stellen, geen “wash out” periode van profylaxe nodig. Momenteel zijn een aantal PK tools of services beschikbaar die nog niet standaard worden gebruikt maar wel publiceren over uitkomsten en resultaten (WAPPS portal, myPKfit, OPTI-CLOT onderzoeksgroep). In Nederland is afgesproken om voor de overgang naar langerwerkende stollingsfactorconcentraten, gebruik te maken van het switch protocol dat een individueel PK profiel op het standaard halfwaardetijd product en het langerwerkend concentraat voor staat (Nederlof, 2018).
Emicizumab (Hemlibra®) is een recombinant gehumaniseerde bispecifieke antistof met zowel affiniteit voor factor IX als factor X waardoor het de functie van factor VIII kan overnemen. Belangrijke positieve eigenschappen van emicizumab zijn de grote biologische activiteit na subcutane toediening, de lange halfwaardetijd van circa 30 dagen en het ontbreken van kruisreactiviteit met factor VIII zodat de aanwezigheid van remmende antistoffen tegen factor VIII geen nadelig effect op de werking heeft. Emicizumab kan worden toegediend als profylaxe bij hemofilie A patiënten mét en zonder anti-factor VIII antistoffen.
Hemofilie A met anti-factor VIII antistoffen
In de HAVEN 1, een fase I studie, werden 109 volwassenen patiënten met ernstige hemofilie A met remmende antistoffen geïncludeerd (Oldenburg, 2017). Met eenmaal per week een subcutane toediening emicizumab werd een 87% reductie gezien van de annualized bleeding rate (ABR) t.o.v. patiënten zonder profylaxe. In vergelijking met patiënten die hieraan voorafgaand profylaxe met bypassing agents kreeg was dit 79%. In totaal was 63% van de patiënten geheel bloedingsvrij.
In de HAVEN 2 studie, een fase III studie in 57 kinderen met ernstige hemofilie A met remmende antistoffen (Young, 2017), werd een ABR reductie van 99% gezien in vergelijking met patiënten met voorafgaande profylaxe met bypassing agents. In totaal was 87% van de patiënten geheel bloedingsvrij.
Hemofilie A zonder anti-factor VIII antistoffen
In de HAVEN 3, een fase III studie, werden 152 patiënten ≥ 12 jaar met ernstige hemofilie A zonder remmende antistoffen geïncludeerd (Mahlangu, 2018). Patiënten die vooraf op on demand therapie stonden werden gerandomiseerd naar subcutaan emicizumab 1,5 mg/kg/week (groep A, N=36) of 3,0 mg/kg elke 2 weken (groep B, N=35) of geen profylaxe (groep C, N=18). Patiënten die vooraf profylactisch werden behandeld, kregen emicizumab 1,5 mg/kg/week (groep D, N=48). De ABR was 1,5 (CI 0,9-2,5) in groep A, 1,3 (CI 0,8-2,3) in groep B en 38,2 (CI 22,9-63,8) in groep C. Het percentage patiënten dat bloedingsvrij was bedroeg 56% in groep A en 60% in groep B. In de 48 patiënten in groep D was de ABR 68% lager met emicizumab dan tijdens factor VIII profylaxe. Er waren geen trombotische events, geen aanwijzingen voor microangiopathie en geen anti-drug antistoffen gevonden.
De HAVEN 4 is een fase III studie in patiënten ≥ 12 jaar met ernstige hemofilie A met en zonder remmers die een subcutaan doseerschema van 6,0mg/kg elke 4 weken heeft onderzocht in 41 patiënten (Pipe, 2019). De mediane ABR voor emicizumab was 4,5 (95% CI 3,1-6,6) en voor behandelde bloedingen 2,4 (1,4-4,3). Na 24 weken behandeling daalde dit naar 2,1 (0,0-5,9) en 0,0 (0,0-2,1) respectievelijk. In totaal was 29% van de patiënten geheel bloedingsvrij en had 56% van de patiënten geen behandeling voor bloedingen nodig.
De HAVEN 6 is een fase III studie in 72 kinderen en volwassenen met milde (n=21) of matig-ernstige (n=51) hemofilie A zonder remmers, bij wie er indicatie gesteld was voor profylaxe. Het betrof 3 vrouwen en 69 mannen. Van deze studie zijn alleen nog interim data, in abstract vorm, beschikbaar (Hermans, 2022, ISTH 2022 congress, OC 30.5). Tijdens een mediane follow-up van 55,6 weken werden geen trombotische of andere relevante complicaties gezien. De model-based ABR (“all bleeds”) in de 24 weken voor de studie was 10.1 (6.9-14.8), welke daalde naar 2.3 (1.7-3.1) tijdens behandeling met emicizumab. De model-based ABR voor “treated bleeds” (de primaire uitkomstmaat in de studie) tijdens emicizumab was 0.9 (0.6-1.5). De meeste van deze bloedingen (79%) waren traumatisch. In totaal had 67% van de patienten geen enkele behandelde bloeding in de studieperiode.
Veiligheidsaspecten
In de HAVEN 1 studie werden 5 patiënten met een trombose of trombotische microangiopathie (TMA) beschreven. Deze waren allen gelijktijdig behandeld met een geactiveerd protrombine complex (aPCC/FEIBA®) ≥ 100 IU/kg/dag ≥ 24 uur. In patiënten met minder aPCC/FEIBA® of alleen rFVIIa (Novoseven®) werden geen trombotische events gezien.
De laatste safety update op 16 oktober 2019, liet in totaal 4 gevallen van TMA zien. Er werden toen meer dan 5200 mensen met hemofilie A met of zonder remmers wereldwijd behandeld. In totaal zijn er 9 gevallen van trombose gemeld: 2 uit de HAVEN 1 en 7 nieuwe gevallen zonder bypassing agents gebruik. In 2 van de 7 gevallen betrof het een longembolie en ischemisch CVA bij verworven hemofilie A; in de 5 andere gevallen een myocardinfarct. Een relatie met emicizumab werd niet aangetoond.
Chirurgie
Er is beperkte ervaring (n=29) met operatieve ingrepen in patiënten met remmende antistoffen die met emicizumab worden behandeld. Het lijkt haalbaar om operaties uit te voeren met lage doses rFVIIa en kleine chirurgie zelfs uit te voeren zonder extra maatregelen (Santagostino, 2019).
In patiënten zonder remmende antistoffen is het mogelijk om factor VIII toe te dienen volgens normale protocollen met normale doseringen onder monitoring met boviene chromogene factor VIII bepalingen.
Monitoring
Emicizumab stoort de one-stage factor VIII bepaling en daardoor ook de reguliere Bethesda assay.
Voor remmende antistof bepalingen moet gebruikt gemaakt worden van een chromogene Bethesda assay met boviene component. Om factor VIII activiteit te meten (residuaal of na suppletie) wordt geadviseerd gebruik te maken van een chromogene factor VIII assay met boviene componenten.
Referenties
Bewijskracht literatuur
Level 1 en Level 3
Zoeken en selecteren
Er werd voor deze uitgangsvraag geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van gepubliceerde richtlijnen (Fischer, 2016; Nordic Haemophilia Guideline 2015; Richards, 2010), aangevuld met bij de auteurs bekende relevante studies.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Geen evidence tabellen, omdat er geen systematisch literatuuronderzoek werd verricht.
Overwegingen
In Nederland is al langer in het algemeen voorkeur voor recombinante stollingsfactoren. Door marktontwikkelingen zijn inmiddels de plasmaproducten duurder dan de recombinante. Dit heeft er toe geleid dat in de afgelopen jaren patiënten die wegens persoonlijk belangrijke redenen nog plasmaproducten gebruikten, bijna allen overgegaan zijn op recombinant. Vanuit de behandelaren gezien is dit een wenselijke situatie.
De enige groep voor wie plasmaproducten overwogen zouden kunnen worden, zijn PUPs en jonge kinderen met ernstige hemofilie A. Er is in Nederland discussie geweest over de externe validiteit en generaliseerbaarheid van de SIPPET studie. Twijfel hierover heeft ertoe geleid dat behandelaren de data bespreken met ouders maar hen niet actief adviseren een plasmaproduct te kiezen. Als na shared decision making besloten wordt tot gebruik van een plasmaproduct, kies dan voor een von Willebrand factor bevattend factor VIII-concentraat.
Omdat er uit de literatuur aanwijzingen zijn dat niet alle recombinante stollingsfactorconcentraten een even groot risico geven op remmer ontwikkeling bij PUPs, wordt aanbevolen om dergelijke patiënten te behandelen met een concentraat dat in deze setting is getest. Of bepaalde producten de voorkeur hebben is niet duidelijk. Beschikbare data uit registraties laten een grote onzekerheidsmarge zien. De significante stijging in remmervorming bij PUPs op de tweedegeneratie producten is niet opgenomen in een aanbeveling, aangezien deze producten in Nederland van de markt gaan. Ten tijde van schrijven van deze richtlijn werd in de meeste centra gekozen PUPs in de eerste 50 exposuredagen te behandelen met een product waarvoor PUP data beschikbaar zijn.
Voor eerder behandelde patiënten zijn er geen aanwijzingen dat preparaten verschillen in immunogeniciteit. Er zijn evenmin aanwijzingen dat switchen een verhoogd risico geeft op remmers. Bij de keuze voor een stollingsfactorconcentraat kan dus prijs en beschikbaarheid een rol spelen. Er zijn geen medische bezwaren tegen het herhaaldelijk wisselen van preparaat. Het is wel vanuit patiënten perspectief onwenselijk om vaker dan eenmaal per 3-5 jaar te wisselen. Wisselingen in product moeten goed begeleid worden, wat extra inzet van personeel nodig maakt.
Bij gebruik van B-domain deleted recombinant factor VIII en bij gebruik van concentraten (zowel factor VIII als IX) met verlengde halfwaardetijd is de one-stage factor VIII of IX bepaling niet altijd bruikbaar. Dit vergt dat centra met hun laboratorium afstemmen welke bepaling past bij de concentraten die gebruikt worden, waarvoor, conform de HKZ richtlijnen, 24×7 een spiegelbepaling beschikbaar zijn.
Langerwerkende stollingsfactoren kunnen worden ingezet om met gelijkblijvende dalspiegels de toedieningsfrequentie te verlagen of om met gelijkblijvende toedieningsfrequentie hogere dalspiegels te bereiken. Verlagen van toedieningsfrequentie is relevant bij patiënten die problemen hebben met veneuze toegang of die hulp nodig hebben bij de toedieningen. In de praktijk is de toename in halfwaardetijd van factor VIII-concentraat vaak niet groot genoeg om minder vaak te kunnen toedienen, bij factor IX lukt dit wel goed. Bereiken van hogere dalspiegels is relevant bij patiënten met een verhoogd bloedingsrisico en bij patiënten die een hoge bloedingsfrequentie hebben ondanks adequate profylaxe met een regulier product. De keuze voor een langerwerkend product is ook afhankelijk van de beschikbaarheid binnen het centrum. Wees bewust dat een lagere frequentie van piek waardes ook nadelig kan zijn voor de patiënt.
Voor het overzetten van standaard- naar verlengde halfwaardetijd producten verwijzen we naar het protocol van de UKCHDO (Collins, 2016).
Binnen Nederland wordt actief onderzoek gedaan naar PK-gestuurd doseren van zowel standaard -halfwaardetijd als langerwerkende stollingsfactorconcentraten. Het is aannemelijk dat hiermee doelmatiger gedoseerd kan worden, daarom wordt ook buiten studieverband al geadviseerd om farmacokinetische parameters mee te nemen bij het doseren van stollingsfactorconcentraat en bij het switchen naar bijvoorbeeld langerwerkende stollingsfactorconcentraten (switch protocol). Bij PK-gestuurd doseren zijn zowel de piek- als de dalwaarde van factor VIII en factor IX van belang. Ook zijn de gebruikte assays (one stage, chromogeen) bij ontwikkelen van het populatie PK model en het individuele PK profiel van groot belang. Hoewel veelbelovend wordt vooral uitgekeken naar ontwikkeling van toekomstige PK-farmacodynamische populatie modellen wegens een betere correlatie tussen factor VIII of factor IX en bloedingsrisico. Gebruikersvriendelijke PK tools en services zijn ontwikkeld of beschikbaar maar worden nog niet breed toegepast.
Het gebruiksgemak (eenmaal per week tot eenmaal per 4 weken subcutaan) en de effectiviteit (vergelijkbaar met een milde hemofilie patiënt met een factor VIII van 30IE/dL) maakt emicizumab een aantrekkelijk alternatief voor factor VIII suppletie. Trombo-embolische risico’s (myocardinfarct) lijken in niet-remmer patiënten voor te komen in patiënten met een verhoogd cardiovasculair risico. Het is onbekend wat er met de tolerantie voor factor VIII gebeurt, wanneer exclusief met emicizumab wordt behandeld; ook is niet bekend of er een reactivatie van remmeractiviteit kan plaatsvinden na re-exposure met factor VIII bij bloedingen of ingrepen.
Toediening van factor VIII naast emicizumab is goed mogelijk met dezelfde dosering als in bestaande protocollen (MASAC recommendation, 2018).
Wij adviseren een geleidelijke introductie van emicizumab in patiënten met congenitale hemofilie A en een indicatie voor profylaxe, ongeacht de ernst van de hemofilie en inclusief draagsters met laag factor VIII, en die: slecht perifeer te prikken zijn, of door andere redenen niet in staat zijn zelf profylaxe toe te dienen, niet goed bloedingsvrij te krijgen zijn met reguliere profylaxe, een zeer actief leven hebben waarbij met reguliere profylaxe onvoldoende bescherming kan worden geboden (b.v. sporters of bij veelvuldig verblijf in buitenland).
Ondanks dat weinig data uit deze leeftijdsgroep beschikbaar zijn, wordt toenemend en met goed resultaat emicizumab toegediend bij kinderen jonger dan 2 jaar. Dit geldt ook voor previously untreated patients (PUPs). In de HAVEN 2 studie die verricht werd in kinderen <12 jaar, waren slechts 9 van de 85 (9.1%) geincludeerde kinderen < 2 jaar (Youing, 2019). In de vergelijkbare HOHOEMI studie waren 1 van 6 (16.7%) en 2 van 7 (28.6%) van de twee studie cohorten < 2 jaar (Shima, 2019). Momenteel lopen er meerdere studies naar gebruik van emicizumab in PUPs of minimaal behandelde jongere kinderen (Young, 2021) (NCT04030052, NCT04431726, NCT04303559). Wij adviseren emicizumab aan te bieden aan kinderen < 2 jaar en daarbij uitgebreid met ouders/verzorgers stil te staan bij de voordelen (subcutane toediening waardoor jonger gestart kan worden met profylaxe en bescherming), onzekerheden en potentiële nadelen van emicizumab toediening in PUPs (geen opbouw tolerantie factor VIII, geen bewezen bescherming tegen intracraniële bloedingen) en in shared decision making een keuze te maken.
Hoe is de behandeling bij een bloeding bij hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Bloedingen zijn het belangrijkste klinische kenmerk bij patiënten met hemofilie en verantwoordelijk voor het merendeel van de morbiditeit en mortaliteit in deze patiënten categorie. Het aantal bloedingen en de ernst daarvan kan per individu sterk variëren.
Onder lichte bloedingen worden verstaan bijvoorbeeld: beginnende hemartros, neus- en tandvleesbloedingen, milde hematurie en bloeding na gering trauma. Ernstige bloedingen zijn bijvoorbeeld: voortgeschreden of ernstige gewrichtsbloedingen, spierbloedingen in boven- en onderarmen, kuit en musculus iliopsoas en ernstige traumata zonder manifeste bloeding. Onder levensbedreigende bloedingen worden verstaan bijvoorbeeld: schedeltrauma, tractus digestivus bloedingen, buiktrauma en bloedingen met bedreiging van de luchtweg.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
Spierbloedingen en gewrichtsbloedingen zijn de meest voorkomende bloedingen bij hemofilie patiënten. Een bloeding in een spier of gewricht kan leiden tot onherstelbare weefselschade (Srivastava, 2013; Britton, 1974; Allain, 1979; Aronstam, 1980) |
|
C |
Een gewrichtsbloeding komt vooral voor in perifere synoviale gewichten zoals de knieën, enkels en ellebogen (Hanley, 2017) |
|
C |
Een spierbloeding van de m. iliopsoas kan leiden tot compressie van de n. femoralis en heeft een hoge recidiefkans. De incidentie van deze bloeding is het hoogst bij jongeren tussen de 10 en 20 jaar oud (Beyer, 2010) |
|
C |
Pasgeboren kinderen met hemofilie hebben de hoogste prevalentie van intracraniële bloedingen (3,5 – 4,0%). Schedeltrauma is de belangrijkste oorzaak voor een intracraniële bloeding, variërend van 38 – 67% (Antunes, 2003; Ljung, 2008; Stieltjes, 2005; Eyster, 1978; Nelson, 1999) |
|
C |
Intracraniële bloedingen in hemofilie patiënten gaan gepaard met een hoge mortaliteit. Behandeling met stollingsfactorconcentraten heeft de mortaliteit ten gevolge van intracraniële bloedingen verlaagd van 70% naar 30% (Antunus, 2003; Stieltjes, 2005; Eyster, 1978; de Tezanos Pinto, 1992; Nuss, 2001; Andes, 1984; Martinowitz, 1986) |
|
A |
In een beperkt aantal oudere studies is er een beneficieel effect van systemisch toegediende tranexaminezuur aangetoond in de preventie van postoperatieve bloedingen bij patienten met hemofilie (van Galen, 2019) |
|
C |
De kans op een gastro-intestinale bloeding neemt toe bij het gelijktijdig gebruik van NSAIDs (Eyster, 2007) |
|
C |
Gastro-intestinale bloedingen komen bij 25% van de hemofilie patiënten voor en zorgen voor een hogere kans op een recidief. De incidentie van gastro-intestinale bloedingen is 10 maal hoger bij hemofilie patiënten dan in de gezonde populatie (Forbes, 1973; Eyster, 2007; Lanas, 2005) |
|
C |
Intra-oculaire bloedingen kunnen leiden tot blijvende visusachteruitgang door beschadiging van de oogzenuw (Rubenstein, 1966) |
Samenvatting literatuur
Resultaten
Algemeen
Er zijn geen gerandomiseerde studies verricht die de behandeling met stollingsfactoren van individuele bloedingen bij patiënten met hemofilie hebben onderzocht. De data die deze aanbevelingen ondersteunen komen voort uit klinische ervaring, ongecontroleerde observationele studies en consensus van expert opinions. Internationale richtlijnen beschrijven hoe om te gaan bij behandeling van bloedingen bij patiënten met hemofilie, formuleren subgroepen van licht, ernstig en levensbedreigend.
Richtlijnen van de United Kingdom Haemophilia Centre Doctors (UKHCDO) stellen dat de maximale tijd tussen de klinische beoordeling van de eventuele aanwezigheid van een bloeding bij presentatie op de SEH het liefste niet langer van 15 minuten mag zijn, en in geval van een bloeding of een verdenking daarop de tijd tot toediening van medicatie niet langer dan 30 minuten mag zijn. De World Federation of Hemophilia (WFH) stelt verder dat de totale duur vanaf het ontstaan van een bloeding tot het toedienen van stollingsmedicatie binnen 3 uur moet plaatsvinden. Tevens geldt het principe: bij twijfel op de aanwezigheid van een bloeding altijd behandelen.
Gewrichtsbloedingen
De UKHCDO richtlijnen beschrijven de kenmerken van en het beleid bij acute gewrichtsbloedingen bij patiënten met hemofilie A en B, zoals hieronder weergegeven (Hanley, 2017).
Symptomen en tekenen van gewrichtsbloedingen (Hanley, 2017).
|
Gewrichts-bloeding |
Symptomen |
Bevindingen bij onderzoek |
Referentie |
|
Milde, beginnende bloeding |
vol gevoel, stijfheid, ongemak, pijn of tintelingen aan het eind van bewegingsuitslag van een gewricht of bij belasten in afwezigheid van een trauma. |
normale beweging |
(Srivastana, 2013; Britton, 1974) |
|
Matige bloeding |
pijn |
enige zwelling, bewegingsbeperking |
(Allain, 1979)
|
|
Ernstige bloeding |
ernstige pijn |
duidelijke zwelling, vrijwel volledige bewegingsbeperking |
(Aronstam, 1980) |
Er is sprake van verdenking op een gewrichtsbloeding bij de volgende symptomen: als eerste functie- en bewegingsbeperking in alle richtingen, daarna zwelling, pijn en warmte. Gewrichtsbloedingen komen vooral voor in de perifere synoviale gewrichten (knieën, enkels en ellebogen). Het is van belang zo snel mogelijk na het ontstaan van een bloeding te starten met de behandeling, omdat de aanwezigheid van bloed in het gewricht leidt tot schade van het gewricht.
Studies in diermodellen laten zien dat door het belasten van een gewricht met een acute bloeding het ontstaan van bot en kraakbeenschade kan verergeren (Hooiveld, 2004). Een andere dierstudie laat zien dat onbelast bewegen van een gewricht na een gewrichtsbloeding pijn, zwelling en inflammatie vermindert (Souza, 2016). Een balans tussen vroege mobilisatie en ontlasten is belangrijk om negatieve effecten van immobilisatie, zoals spieratrofie en contractuurvorming, te voorkomen, de proprioceptie te verbeteren en optimaal functioneel herstel te bewerkstelligen (Blamey, 2010).
Spierbloedingen
De World Federation Haemophilia (WFH) guidelines beschrijven de huidige aanbevelingen voor het behandelen van spierbloedingen bij patiënten met hemofilie A en B (Srivastava, 2013).
Een intramusculaire spierbloeding kenmerkt zich door pijn en een functionele verkorting van de spier waardoor een functie beperking van de aangrenzende gewrichten ontstaat. Afhankelijk van de locatie van de spierbloeding is er zwelling zichtbaar of palpabel (Beyer, 2010). Echografie kan gebruikt worden om een spierbloeding te diagnosticeren en de resorptie te vervolgen (Querol, 2012). Een spierbloeding van de m. iliopsoas wordt gekenmerkt door flexiestand van de heup en hyperlordose van de lumbale wervelkolom en normale rotaties van de heup. Een spierbloeding van de m. iliopsoas kan leiden tot compressie van de n. femoralis en uitval van de m. quadriceps functie en heeft een hoge recidiefkans (Beyer, 2010).
Intracraniële bloedingen
Een intracraniële bloeding bij hemofilie patiënten is de meest gevreesde bloedingscomplicatie en gaat gepaard met een hoge mortaliteit (Antunes, 2003; Stieltjes, 2005; de Tezanos Pinto, 1992; Nuss, 2001; Andes, 1984). Pasgeboren hebben de hoogste prevalentie (3,5 – 4,0%) (Ljung, 2008). Risicofactoren voor intracraniële bloedingen zijn de ernst van de hemofilie, jonge leeftijd (pasgeborenen) en een leeftijd boven de 50 jaar, aanwezigheid van een remmer, HIV en hepatitis C infectie (Nuss, 2001; Fransen van de Putte, 2012). Behandeling met stollingsfactorconcentraten heeft de mortaliteit van hemofilie patiënten met een intracraniële bloeding verlaagd van 70% naar minder dan 30% (Eyster, 1978; Martinowitz, 1986). Hoe eerder de toediening van stollingsfactorconcentraten plaatsvindt bij een bloeding in het centraal zenuwstelsel, des te sneller een bloeding tot staan gebracht wordt met afname van morbiditeit en mortaliteit.
Het advies ten aanzien van de streefspiegel van factor VIII of factor IX na een schedeltrauma of spontane bloeding van het centraal zenuwstelsel is afhankelijk van de aard van het trauma en localisatie van de bloeding. Traivaree onderstreept de noodzaak van het verrichten van een CT scan bij een patiënt met een ernstige hemofilie die onverklaarde neurologische symptomen heeft en een hoge verdenking op een bloeding in het centrale zenuwstelsel (Traivaree, 2007).
Gastro-intestinale bloedingen
Bloedingen uit het bovenste gedeelte van het maag-darmkanaal (slokdarm, maag en twaalfvingerige darm) komen voor bij 25% van patiënten met hemofilie en hebben een hoge recidief kans (Forbes, 1973). Een multicenter studie door Eyster et al in 2007 toonde een jaarlijkse incidentie van 1,3% op bovenste gastro-intestinale bloedingen bij hemofilie patiënten (Eyster, 2007). Deze incidentie is 10 maal hoger dan vergeleken met de gezonde populatie (Lanas, 2005). In dezelfde studie van Eyster werd gevonden dat de kans op een bovenste gastro-intestinale bloeding toenam bij het gelijktijdig gebruik van NSAID’s binnen één maand.
Patiënten met hemofilie hebben ook een verhoogde kans op het ontwikkelen van een gastro-intestinale bloeding met daarna een hogere kans op een recidief (Lanas, 2005). Deze kans neemt verder toe bij het oplopen van de leeftijd (>46 jaar) en bij leverdecompensatie, gedefinieerd als het optreden van ascites, varices, hepatitis C, variceuze bloeding of lever encefalopathie (Eyster, 2007). Het gebruik van NSAID’s gedurende minder dan een maand, geeft al voor een toegenomen kans op een gastro-intestinale bloeding bij hemofilie (Eyster, 2007). Corticosteroïden verhogen ook de kans op een gastro-intestinale bloeding, zeker bij gelijktijdig gebruik met NSAID’s, (Nielsen, 2001).
Hematurie
Bij patiënten met hemofilie komt hematurie vaak voor, in twee studies werd een levenslange incidentie tot 66% beschreven (Beck, 1972; Prentice, 1971). Er zijn geen gerandomiseerde studies verricht die de behandeling van hematurie bij patiënten met hemofilie hebben onderzocht (Srivastava, 2013).
Oogbloedingen
Er zijn geen gerandomiseerde studies verricht die de behandeling onderzoeken van intra-oculaire bloedingen bij hemofilie A of B patiënten. Intra-oculaire bloedingen kunnen leiden tot blijvende visusachteruitgang door compressie van de oogzenuw (Rubenstein, 1966). Symptomen die kunnen duiden op een oogbloeding zijn een snelle, soms pijnlijke plotseling achteruitgang van de visus spontaan of na een hoofdtrauma. Een trauma aan het oog betreft vooral het voorste oogsegment waardoor hyphema kan ontstaan (Wang, 2008).
Referenties
Bewijskracht literatuur
Level 3, consistent
Zoeken en selecteren
Gewrichtsbloedingen
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van 3 eerder gepubliceerde evidence-based richtlijnen. Vervolgens werd een literatuursearch verricht vanaf 2017, het jaar van de meest recente guideline voor aanvullende literatuur met de volgende zoektermen: “Hemophilia”, “joint bleed” and “treatment”, met restrictie “inhibitors”. Dit resulteerde in 44 publicaties. Na titel en abstract screening werden 4 publicaties geselecteerd voor selectie op basis van de volledige tekst, waaronder 1 review. De overige publicaties werden geëxcludeerd op basis van het ontbreken van relevante informatie over de vraag (n=40). Uit de geselecteerde artikelen werden de referenties gescreend op additionele relevante artikelen. Dit leverde nog 4 artikelen op.
Spierbloedingen
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van 3 eerder gepubliceerde evidence-based richtlijnen. Vervolgens werd een literatuursearch verricht vanaf 2012, het jaar van de meest recente guideline, voor aanvullende literatuur met de volgende zoektermen: “Hemophilia” AND “muscle bleed” OR “muscle hematoma”. Dit resulteerde in 78 publicaties. Na titel en abstract screening werden 4 publicaties geselecteerd voor selectie op basis van de volledige tekst, waaronder 1 review. De overige publicaties werden geëxcludeerd op basis van het ontbreken van relevante informatie over de vraag (n=74). Uit de geselecteerde artikelen werden de referenties gescreend op additionele relevante artikelen. Dit leverde geen extra artikelen op.
Intracraniële bloeding
Er werd voor de ze uitgangsvraag een systematische review verricht met de volgende zoektermen: “Hemophilia” and “cerebral trauma” or “cerebral hemorrhage” or “intracranial bleeding / hemorrhage” zonder restricties. Dit resulteerde in 380 publicaties waarvan er 308 na ontdubbelen overbleven. Na titel en abstract screening resulteerde dat in 111 relevante publicaties, waaronder veel case reports, retrospectieve studies en reviews.
Gastro-intestinale bloedingen
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht.
Hematurie
Er werd voor deze uitgangsvraag een systematische review verricht met de volgende zoektermen: “Hemophilia” and “hematuria” zonder restricties. Dit resulteerde in 204 publicaties. Na titel en abstract screening resulteerde dat in 46 relevante publicaties, waaronder veel case reports, retrospectieve studies en reviews.
Oogbloeding
Er werd voor deze uitgangvraag een systemische review verricht met de volgende zoektermen: “Ophthalmological bleeding” or “eye bleeding” and “Hemophilia” zonder restricties. Dit resulteerde in een beperkt aantal publicaties (35). Na titel en abstract screening resulteerde dat in 6 relevante publicaties.
Zoekverantwoording
De literatuursearch werd verricht in de volgende elektronische databases: Pubmed, Cochrane en MEDLINE. Na ontdubbelen zijn de publicaties gecontroleerd op taal (Engels en Nederlands) en relevantie (case reports, observationele en retrospectieve studies).
Overwegingen
In de formule over continue infusie factor VIII en IX wordt de gewenste infusiesnelheid in IE/kg/uur berekend. Er is voor gekozen om geen vaste infusie hoeveelheid aan te bevelen, omdat het aantal eenheden stollingsfactor afhangt van de activiteitswaarde die wordt nagestreefd (zoals aanbevolen in de tabellen 6.1 en 6.2). In de formule wordt de streefwaarde stijging vermenigvuldigd met de klaring. De streefwaarde stijging is het verschil tussen de gewenste streefwaarde minus de eigen basale factor VIII/IX activiteit in IE/dL.
De klaring is een getal dat is afgeleid uit de data afkomstig uit de Nederlandse OPTI-CLOT trial, een multicenter Nederlands onderzoek naar perioperatieve substitutie therapie bij hemofilie.
In het model voor factor VIII (Hazendonk, 2016):
In model voor factor IX (Preijers 2018):
In de OPTI-CLOT trial data (factor VIII) zijn de volgende klaringen gezien wanneer de samples pre-operatief en net postoperatief worden meegenomen: mediaan (range): 0,024 (0,012 – 0,059) dL/h/kg (leeftijd range 4-76 jaar).
Gezien deze uitkomsten wordt er onderscheid gemaakt in factor VIII en IX en in volwassenen en kinderen, zoals aangegeven in de aanbevelingen.
Tabel 6.1 Algemene behandeling van bloedingen bij hemofilie A+ en B# naar ernst van de bloeding
|
Ernst van de bloeding |
Initiële top streefspiegel factor VIII/IX (IE/dl) |
Streefdal spiegel I.T. onderhoud factor VIII/IX (IE/dl) |
Streefspiegel C.I. factor VIII/IX (IE/dl) |
Gebruikelijke minimale duur (dagen) |
|
mild* |
30 |
– |
– |
– |
|
ernstig** |
50 |
>25 |
30 – 50 |
2-7 |
|
levens- bedreigend *** |
100 – – |
>50
|
80 – 100 50 – 80 30 – 50 |
1, hierna 2 – 6 7 – 14 |
I.T. = intermitterende therapie; C.I. = continue infusie
+ Bereken de dosis stollingsfactorconcentraat voor de intermitterende behandeling met de formule bij hemofilie A: (streefplasma spiegel in IE/dl – pre-existente spiegel in IE/dl) / 2 X gewicht in kg
# Bereken de dosis stollingsfactorconcentraat voor de intermitterende behandeling met de formule bij hemofilie B: (streefplasma spiegel in IE/dl – pre-existente spiegel in IE/dl) X gewicht in kg
* mild = bloeding waarbij geen langdurige verhoging van stollingsfactor spiegel wordt aanbevolen zoals epistaxis, beginnende haemartros en tandvleesbloedingen.
** ernstig = bloeding waarbij na een oplaaddosering gedurende meerdere dagen een minimale stollingsfactorspiegel nagestreefd moet worden, zoals een voortschrijdende spier- en gewrichtsbloeding, andere ernstige bloedingen en ernstig trauma zonder manifeste bloeding.
*** levensbedreigend = bloeding waarbij na een oplaaddosering meerdere dagen een minimale stollingsfactorspiegel nagestreefd moet worden i.v.m. een directe bedreiging van een orgaan of hoge kans op verbloeding zoals een intracraniële bloeding, bloeding met bedreiging van de luchtweg.
Tabel 6.2 Richtlijn behandeling van specifieke bloedingen bij hemofilie A en B
|
Type bloeding |
|
Initiële top streefspiegel factor VIII/IX (IE/dL) |
Streefdal spiegel I.T. onderhoud factor VIII/IX (IE/dL) |
Streefspiegel C.I. factor VIII/IX (IE/dL) |
Gebruikelijke minimale duur** (dagen) |
|
Gewrichts-bloeding |
vroeg |
30 |
– |
– |
eenmalig |
|
matig |
50 |
>25 |
30-50 |
2 – 5 |
|
|
ernstige |
100 |
>50 |
50-80 |
4-7, hierna afbouw-schema |
|
|
Spierbloeding |
mild |
50 |
>25 |
30-50 |
1-3 |
|
ernstig |
100 |
>50 |
80-100 |
1-2, hierna |
|
|
|
– |
>50 |
50-80 |
3-5 |
|
|
Schedeltrauma
|
mild (val <50cm, lichte klap) |
100 |
– |
– |
eenmalig |
|
matig-ernstig* (val>50cm, forse klap) |
100 |
>50 |
50-80 |
1-3 |
|
|
ernstig* (commotio / contusio) |
100 |
>50 |
80-100 |
3 – 7 |
|
|
Intracraniële bloeding |
|
100 |
>50 |
80-100 |
6, hierna |
|
|
– |
>50 |
50-80 |
7 – 10, hierna |
|
|
|
– |
>30 |
30-50 |
11 – 14 |
|
|
Gastro- intestinale bloeding# |
|
100 |
>80 – 100 |
100 |
1, hierna |
|
|
– |
>50 |
50-80 |
2 – 6, hierna |
|
|
|
– |
>30 |
30-50 |
7 – 10 |
|
|
Hematurie$ |
|
50 |
>30 |
30-50 |
1 – 5 |
|
Oogletsel met bedreigde visus+ |
|
100 |
>50 |
80-100 |
1, hierna |
|
– |
>50 |
50 -80 |
2 – 6 |
||
|
– |
>30 |
30 – 50 |
7 – 14 |
||
|
Epistaxis$ |
|
30 |
– |
– |
eenmalig |
|
Mondslijmvlies, tandvlees bloeding |
|
30 |
– |
– |
eenmalig |
I.T. = intermitterende therapie; C.I. = continue infusie
* strikte monitoring
** sterk afhankelijk van lokalisatie en ernst bloeding, als ook conditie gewricht, leeftijd en activiteiten patroon etc.
# uitgaande van een levensbedreigende gastro-intestinale bloeding
$indien er sprake is van een behandelindicatie
+behandel als levensbedreigende bloeding
Bij kinderen worden i.v.m. een andere farmacokinetiek die zich kenmerkt door een grotere klaring, een groter verdelingsvolume en daardoor een kortere halfwaarde tijd, voor milde bloedingen vaak een initiële top streefspiegel factor VIII/factor IX nagestreefd van 50 IE/dl; bij ernstige bloedingen van een top streefspiegel van 80 IE/dl en zoals ook bij volwassenen bij een levensbedreigend bloeding een top streefspiegel van 100 IE/dl. Frequente monitoring in kinderen is van belang ter bewaking van top en ook dalspiegels.
Gewrichtsbloedingen
Bij een beginnende hemartros is over het algemeen eenmalige toediening van stollingsfactoren voldoende effectief. Een beginnende hemartros wordt gekenmerkt door een vol gevoel, stijfheid, ongemak, pijn of tintelingen aan het eind van bewegingsuitslag van een gewricht in afwezigheid van een trauma. Daarbij is er nog geen bewegingsbeperking van het gewricht vast te stellen. Bij een matig- ernstige bloeding, waarbij er wel enige zwelling en bewegingsbeperking is, zijn 2 toedieningen van stollingsfactoren meestal voldoende. Follow up van therapeutisch effect is altijd geïndiceerd. In geval van acute hemartros gepaard gaande met ernstige bewegingsbeperking en zwelling, en pijn wordt substitutietherapie gecontinueerd totdat de functie is verbeterd en zwelling duidelijk is afgenomen of gestabiliseerd, therapeutisch of profylactisch. De dosering van de stollingsfactorsuppletie wordt bepaald aan de hand van de ernst van de bloeding. De initiële streef factor VIII/IX plasmaspiegel bedraagt > 30 IE/dl bij beginnende hemartros en bij een matig-ernstige bloeding > 50-60 IE/dl. Bij een ernstige hemartros met bewegingsbeperking > 80-100 IE/dl.
Veel symptomen die geassocieerd worden met een gewrichtsbloeding komen ook voor bij een opvlamming (exacerbatie) van hemofilie-artropathie, waaronder pijn, bewegingsbeperking en zwelling (Timmer, 2015). Wanneer een gewrichtsbloeding niet kan worden uitgesloten, dient laagdrempelig behandeld te worden met suppletietherapie. Een kleine studie laat zien dat musculoskeletale klachten bij mensen met hemofilie regelmatig incorrect gediagnosticeerd worden (Ceponis, 2013). Het gebruik van point-of-care echografie wordt in twee kleine studies accuraat bevonden voor het detecteren van bloed in een gewricht (Ceponis, 2013; Nguyen, 2018).
Een gewrichtspunctie met aspiratie van bloed is gecontra-indiceerd, tenzij er verdenking is op een potentiële septische artritis. Dan is het noodzakelijk om materiaal te verkrijgen voor diagnostiek en inzetten van een kweek. Bij een ernstige bloeding in het heupgewricht dient decompressie artrocentese te worden overwogen om gewrichtschade door druk op vasculatuur van het caput femoris te verminderen en de kans op kopnecrose te verkleinen. Een gewrichtspunctie met aspiratie kan zinvol zijn voor verlichting van pijn bij een gespannen hemartros onder adequate substitutie therapie. Hierbij moet een streef plasmapiegel > 30-50 IE/dl gedurende 47-72 uur worden aangehouden. Nadien is het van belang het gewricht te immobiliseren, compressie van het gewricht en het gewricht gedurende 24-48 uur niet te belasten.
De positieve invloed van koelen met ijs wordt in twijfel getrokken (Rodriguez-Merchan, 2017). Kleine studies laten zien dat koelen pijnklachten vermindert, inflammatie en zwelling verbetert en de benodigde hoeveelheid stollingsfactorconcentraat vermindert (Ravanbod, 2017). Andere studies in diermodellen en mensen laten zien dat koelen van bloed en/of weefsels stollingsfactor activiteit en trombocytenfunctie kan verminderen en bloeden juist kan verlengen (Forsyth, 2012).
Voor pijnbestrijding kan er gekozen worden voor stapsgewijs voorschrijven van de volgende medicatie: paracetamol, COX-2 selectieve NSAIDs en opioïden.
Het belasten van een gewricht met een gewrichtsbloeding kan het ontstaan van bot en kraakbeenschade verergeren. Bij een acute gewrichtsbloeding, dient het gewricht ontlast te worden door immobilisatie met behulp van krukken of een sling. Patiënt dient goed geïnstrueerd te worden hoe deze hulpmiddelen optimaal gebruikt kunnen worden. Het immobiliseren van een gewricht met behulp van een spalk wordt niet routinematig aanbevolen, maar kan overwogen worden bij een jong kind (dat moeilijk te immobiliseren is) met een grote gewrichtsbloeding. Hierbij mag er geen circulair gips aangemeten worden. Wanneer de bloeding gestopt is (zwelling, warmte en pijn nemen af, bewegingsuitslag neemt toe) wordt de revalidatie gestart. Een balans tussen vroege mobilisatie en ontlasten is belangrijk om negatieve effecten van immobilisatie te voorkomen en optimaal functioneel herstel te bewerkstelligen (ook van de propriocepsis). Revalidatie is initieel gericht op gewrichtsfuncties (bewegingsuitslag, stabiliteit, kracht). Passieve mobilisatie en interventies die het hemostatische herstelproces kunnen verstoren, zoals massage of warmteapplicatie, zijn in de subacute fase na een bloeding gecontra-indiceerd. Bij herstel van de bewegingsuitslag wordt de focus verlegd naar functionele revalidatie (gericht op activiteiten en participatie). Wanneer de bewegingsbeperking voldoende is hersteld, kan functionele revalidatie worden gestart. Functionele revalidatie wordt beëindigd als het activiteitniveau volledig hersteld is en re-integratie plaats heeft gevonden. Patiënteducatie met betrekking tot het identificeren en behandelen van een gewrichtsbloeding dient tijdens het gehele traject plaats te vinden.
Spierbloedingen
Spierbloedingen zijn, na gewrichtsbloedingen, de meest voorkomende bloedingen. Bij ernstige hemofilie zijn 10-15% van de bloedingen spierbloedingen. Een spierbloeding kan inter-of intramusculair zijn. Een intramusculaire spierbloeding kenmerkt zich over het algemeen door pijn en een functionele verkorting van de spier met een functiebeperking van de aangrenzende gewrichten. Op lengte brengen en aanspannen van de spier is pijnlijk. Afhankelijk van de locatie van de spier is een gespannen zwelling zichtbaar en is er vaak een lokale verharding palpabel. Bij diepliggende (houdings-)musculatuur is er geen zwelling zichtbaar of palpabel. Na enkele dagen kan een hematoom zichtbaar worden. Een spierbloeding wordt gediagnosticeerd op basis van anamnese en lichamelijk onderzoek, daarnaast kan aanvullend onderzoek (echo, MRI, CT) geïndiceerd zijn.
De behandeling van een intramusculaire spierbloeding bestaat in eerste instantie uit het toedienen van stollingsfactoren en het ontlasten van de spier met behulp van krukken of een sling, bij een ernstige spierbloeding bedrust. Compressie bij een spierbloeding wordt afgeraden, dit kan zorgen voor een verhoging van de intramusculaire druk met het risico op compressie van een zenuw een compartimentsyndroom. Als de bloeding gestopt is (pijn neemt af, zwelling neemt af en bewegingsuitslag neemt toe) kan er gestart worden met actief mobiliseren binnen de pijngrens. Op rek brengen van de spier moet in deze fase voorkomen worden. Oefentherapie is daarom altijd actief en gericht op het aanspannen van de antagonisten. Als de volledige bewegingsuitslag hersteld en pijnvrij is kan functionele revalidatie gestart worden. De duur tot volledig herstel is afhankelijk van de grote van de bloeding, en kan tot 6 maanden duren. Met behulp van echografie kan de resorptie van het hematoom vervolgd worden. De mate van resorptie kan gebruikt worden als leidraad voor revalidatie en opbouwen van activiteiten.
Een spierbloeding die veel gezien wordt bij jongeren tussen 10 en 20 jaar is de m. iliopsoas bloeding. De zwelling is bij deze spier moeilijk zichtbaar of palpabel. De functionele spierverkorting zorgt echter voor een flexiestand van de heup en vergrote lordose in de lumbale wervelkolom. De rotaties van het heupgewricht zijn normaal. Daarnaast wordt er pijn aangegeven in de lage rug en liesregio. Een bloeding van de m. iliopsoas moet onderscheden worden van een bloeding in het heupgewricht (hierbij zijn de rotaties beperkt) en problematiek van de buikingewanden (denk aan een appendicitis). Bij een m. iliopsoas bloeding kunnen vervelende complicaties optreden, waaronder compressie van de n. femoralis. Dit zorgt voor een verminderde sensibiliteit van de anterior en mediale zijde van het bovenbeen en kan leiden tot uitval van de m. quadriceps. De m. iliopsoas bloeding heeft een hoge recidiefkans, begeleiding tot volledig functioneel herstel is hierbij dus essentieel.
Complicaties die kunnen ontstaan als een spierbloeding niet goed behandeld wordt zijn contractuurvorming, zenuwinklemming, compartimentsyndroom en het ontstaan van een pseudotumor (bij ernstige hemoglobine daling).
Schedeltrauma en intracraniële bloedingen
In verschillende, meest retrospectieve studies, wordt het optreden van een bloeding in het centraal zenuwstelsel bij hemofilie patiënten beschreven met een wisselende incidentie (Antunes, 2003; Ljung, 2008). Dit wordt mede veroorzaakt door de grote verschillen in patiënt karakteristieken binnen de studies m.b.t. o.a. de leeftijd van de onderzoekspopulatie, het bestaan van profylactische behandeling met stollingsfactorconcentraat en het hebben van neutraliserende antistoffen (Nakar, 2010). Er zijn geen gerandomiseerde studies verricht die de duur van behandeling met stollingsfactorconcentraten bij een bloeding in het centraal zenuwstelsel hebben onderzocht. Gerandomiseerde studies die gekeken hebben naar het juiste moment van opstarten van profylactische behandeling stollingsfactorconcentraten bij het optreden van centraal zenuwstelsel bloedingen ontbreken.
Intracraniële bloedingen vormen een potentieel levensbedreigende complicatie van hemofilie A en B, gepaard gaande met een hoge morbiditeit en mortaliteit. Pasgeboren hebben de hoogste prevalentie (3,5 – 4,0%) (Ljung, 2008). Schedeltrauma is de belangrijkste oorzaak van een intracraniële bloeding, waarbij 38 – 67% van intracraniële bloedingen vooraf worden gegaan door een trauma (Antunes, 2003; Stieltjes, 2005; Eyster, 1978; Nelson, 1999). Spontane bloedingen in het centrale zenuwstelsel komen daarnaast ook regelmatig voor bij jonge patiënten met ernstige hemofilie A en B (de Tezanos Pinto, 1992). Intracraniële bloedingen worden gekarakteriseerd over het algemeen door focale neurologische tekenen en insulten, extracerebrale bloedingen leiden vaak tot diffuse hoofdpijn en veranderd bewustzijn (Klinge, 1999).
Spontane bloedingen komen weer vaker voor dan bloedingen na een hoofdtrauma, waardoor alertheid gewenst op een bloeding in het centraal zenuwstelsel indien er onverklaarbare neurologische symptomen optreden zoals plotseling hoofdpijn of pijn in de rug, verminderd bewustzijn of focale uitvalsverschijnselen.
Het advies ten aanzien van de streefspiegel van factor VIII of factor IX na een schedeltrauma of spontane bloeding van het centraal zenuwstelsel is afhankelijk van de aard van het trauma en plaats van de bloeding. Traivaree onderstreept de noodzaak van het maken van een CT scan bij een patiënt met een ernstige hemofilie die onverklaarde neurologische symptomen heeft en een hoge verdenking op een bloeding in het centrale zenuwstelsel (Traivaree, 2007).
Gastro-intestinale bloedingen
De meest voorkomende oorzaak van bloedingen in het bovenste gedeelte van het maagdarmkanaal zijn peptische ulcera (Mittal, 1985), waaraan in het verleden tot meer dan 30% van de hemofilie patiënten door bloedingen overleed. Helicobacter pylori infectie gaat vaak samen met een peptisch ulcus en wordt in dezelfde frequentie in hemofilie patiënten gevonden. Het valt aan te bevelen om hemofilie patiënten die een NSAID gebruiken om te zetten naar een selectieve COX2 remmer, waardoor de kans op een bloeding uit een peptisch ulcus verlaagd wordt. Het gelijktijdig gebruik van een zuurremmer (protonpomp remmer of histamine 2-receptor antagonist) wordt aanbevolen bij het starten van corticosteroïden (Kouides, 2010). Bij levercirrose en portale hypertensie wordt naast het gebruik van een zuurremmer ook een bèta-blokker geadviseerd om het risico op een bloeding uit varices te verlagen (Thalheimer, 2008).
In het geval van een gastro-intestinale bloeding is het van belang de hemodynamiek van een patiënt goed te vervolgen, waardoor een ziekenhuisopname vaak onontbeerlijk is. Een acute gastro-intestinale bloeding kan zich presenteren als hematemesis, hematochezia en melena. Naast algemeen ondersteunende maatregelen zoals het starten van een zuurremmer en bloedtransfusies, dienen direct factor VIII of factor IX stollingsfactorconcentraten toegediend te worden tot een streefwaarde van 100 IE/dL, die over een periode van 10 dagen afgebouwd kan worden indien de bloeding tot staan is gebracht. Voor het onderzoeken van de oorzaak van de bloeding, wordt vaak een gastroscopie uitgevoerd. Het gebruik van tranexaminezuur naast stollingsfactorconcentraten wordt aanbevolen om de slijmvliesbloeding uit het maagdarmkanaal sneller onder controle te krijgen (Kouides, 2008).
Hematurie
Bloedingen als oorzaak van hematurie kunnen op elk niveau in de urinewegen ontstaan en de differentiaal diagnose is breed (Cohen, 1966). Patiënten met hemofilie hebben een verhoogde kans op nefrolithiasis door de hogere incidentie van osteoporose met een toegenomen excretie van calcium in de urinewegen. Hierbij kan overgewicht en een verminderde actieradius op latere leeftijd ook een rol spelen. Chronische artritis door recidiverende gewrichtsbloedingen leidt tevens tot een toegenomen botresorptie (Ghosh, 2003).
Pijnloze hematurie bij hemofilie patiënten wordt beschouwd als een bloeding door een tekort aan factor VIII dan wel factor IX. Krachtige hydratie met 2-3L vocht per dag laat de bloeding vaak vanzelf verdwijnen (Srivastava, 2013; Quon, 2010). Indien patiënten dit niet kunnen drinken wordt intraveneuze vochttoediening geadviseerd. Er is geen bewijs voor het voordeel van totale bedrust bij hematurie. Daarnaast is deze maatregel in de praktijk bij symptomatische patiënten slecht uitvoerbaar.
Persisterende en pijnlijke hematurie dient tevens behandeld te worden met stollingsfactorconcentraten om de bloeding te kunnen stoppen, waarbij goed gelet moet worden voor het optreden van stolsels en urineweg obstructie (Srivastava, 2013; Quon, 2010; Ghosh, 2003).
DDAVP is gecontra-indiceerd bij hematurie vanwege het advies om bij hematurie geen vochtrestrictie aan te houden maar juist een krachtig hydratie na te streven. Gelijktijdig gebruik van DDAVP kan leiden tot een ernstige hyponatriëmie. Antifibrinolytica bij bloedingen uit de bovenste urinewegen mogen niet toegepast worden in verband met de kans op retentie van stolsels in de ureteren en urineblaas (Mannuci, 1998) hetgeen kan resulteren in mogelijke urinewegobstructie (Pitts, 1986).
Oogbloedingen
Spontane intra-oculaire bloedingen bij patiënten het hemofilie A of B worden zelden in de literatuur beschreven. De meeste oogbloedingen bij hemofilie patiënten treden op na een trauma van het hoofd (Rubenstein, 1966; Maguluri, 2005).
Om blijvende oogzenuwschade en daardoor een blijvende visusbeperking door een bloeding in het oog bij hemofilie A en B patiënten te voorkomen, wordt geadviseerd bij de geringste verdenking op een oogbloeding direct te starten met stollingsfactorconcentraten. Voor dosering en duur van de behandeling zie tabel 6.2 (Srivastatva, 2013; Bush, 1995; Guthrie, 1980; Singleton, 2010). Behandeling met DDAVP is bij een oogbloeding gecontra-indiceerd in verband met een verhoogde conjunctivale injectie waardoor een recidief bloeding kan optreden in de voorste oogkamer.
Objectivering van oogbloedingen geschiedt door middel van oftalmologisch onderzoek door consultatie van de oogarts (Srivastava, 2013). Bij een bewezen bloeding in het oog is de aanbevolen behandelduur 14 dagen.
Behandeling met stollingsfactorconcentraten
Behandeling met stollingsfactorconcentraat is vaak een belangrijk onderdeel van de behandeling van bloedingen. De halfwaardetijd van standaard half-life factor VIII-concentraat bedraagt gemiddeld 8 tot 15 uur. De halfwaardetijd van standaard half-life factor IX-concentraat bedraagt gemiddeld 18 tot 24 uur. De farmacokinetiek van factorconcentraat kan echter per individu sterk verschillen en kan leiden tot noodzakelijke aanpassingen van dit beleid. Op den duur zullen patiënten mogelijk standaard op basis van individuele farmacokinetiek met populatie farmacokinetische modellen worden behandeld. Extended half-life factorconcentraten worden minder snel geklaard. De dosering van deze producten is nog in onderzoek.
Toediening stollingsfactorconcentraten bij bloedingen
De toediening van één eenheid (IE) standaard half-life factor VIII-concentraat per kilogram (kg) geeft een gemiddelde toename van factor van circa 2 IE/dl in het plasma van de patiënt. Een rekenvoorbeeld: indien een plasmaconcentratie van 50 IE/dl is gewenst bij een patiënt met ernstige hemofilie A van 70 kg, moet (70 x 50 IE) /2 = 1750 IE worden geïnfundeerd. Omdat de gemiddelde halfwaardetijd van standaard factor VIII-concentraat ongeveer 12 uur is, dient elke 12 uur de helft van de oplaaddosis toegediend te worden (875 IE) om de plasma factor VIII-concentratie te handhaven boven de 25 IE/dl (dalspiegel). Dit moet echter wel gemonitord worden d.m.v. plasma spiegels i.v.m. de inter-individuele verschillen in farmacokinetiek.
De toediening van één eenheid (IE) standaard half-life factor IX-concentraat per kilogram (kg) geeft een gemiddelde toename van factor van circa 1 IE/dl in het plasma van de patiënt. Een rekenvoorbeeld: indien een plasmaconcentratie van 50 IE/dl gewenst is bij een patiënt van 70 kg met ernstige hemofilie B moet (70 x 50 IE) /1 = 3500 IE worden geïnfundeerd. Omdat de gemiddelde halfwaardetijd van standaard halfwaarde factor IX ongeveer 18-24 uur is, is toediening van de halve oplaaddosis eenmaal per dag (1750 IE) voldoende om de factor IX spiegel tussen 25 en 50 IE/dl te handhaven (dalspiegel). Dit moet echter wel gemonitord worden d.m.v. plasma spiegels i.v.m. de inter-individuele verschillen in farmacokinetiek.
Hoe is de behandeling rondom een ingreep of operatie?
Aanbevelingen
Onderbouwing
Inleiding
Operaties en ingrepen bij hemofilie patiënten moeten onder stollingsfactorcorrectie plaatsvinden om bloedingscomplicaties te voorkomen. Preoperatief wordt een oplaaddosis met stollingsfactorconcentraat gegeven die voldoende is om de gewenste spiegel te bereiken. Het is voor iedere ingreep belangrijk dat lokale hemostase wordt bereikt.
Conclusies
|
SORT Grade |
Conclusie |
|
B |
Voor circumcisie is suppletie met stollingsfactor VIII en IX noodzakelijk: in totaal twee tot vier dagen behandeling met bij voorkeur de eerste 24 uur na de ingreep factor VIII en IX spiegels > 80 IE/dL en 24-96 uur na de ingreep > 50 IE/dL. (Yilmaz 2010, Hermans 2009) |
|
B |
Bij circumcisie is aanvullende behandeling met tranexaminezuur (antifibrinolyticum) noodzakelijk: systemisch oraal of intraveneus, start een dag voor de ingreep en continueren tot 7-10 dagen na de ingreep. (Yilmaz 2010) |
|
C |
Bij circumcisie is fibrin glue op het wondvlak gewenst (Yilmaz 2010). Het is wenselijk het verband van het wondgebied nog tijdens suppletietherapie te verwijderen. |
|
C |
Voor invasieve tandheelkundige ingrepen is het noodzakelijk dat het stollingsfactorgehalte minimaal 50-60 IE/dL is. (Anderson 2013) |
|
B, C |
Bij invasieve tandheelkundige ingrepen is het nodig bij kinderen en volwassenen aanvullend een antifibrinolyticum te geven, bij voorkeur systemisch oraal te starten een dag voor de ingreep tot 7 dagen na de ingreep. Het is wenselijk dit te combineren met tranexaminezuur mondspoeling gedurende de antifibrinolytische therapie. (Anderson 2013) |
|
C |
Correctie van stollingsfactoren is noodzakelijk voor plaatsen van een mandibulair blok en linguale infiltratie van de mondbodem. Bij kinderen kan ook correctie van stollingsfactoren overwogen worden voor buccale infiltratie, intra-papillaire injectie en intraligamentaire injecties. (Anderson 2013) |
|
C |
Voorafgaande aan percutane coronaire interventie (PCI) of coronaire angiografie (CAG) is stollingsfactorcorrectie noodzakelijk, naar een piekspiegel van 80 IE/dL voorafgaand, en dalspiegels van 50 IE/dL tot 24 uur na de interventie uitgaande van een a. radialis benadering. Het is noodzakelijk hogere stollingsfactor VIII en IX piekspiegels dan 80 IE/dL te vermijden om occlusieve thrombi te voorkomen. (Schutgens 2009, Tuinenburg 2013) |
|
A |
Het is noodzakelijk de arteria radialis te gebruiken als toegangsweg voor percutane coronaire interventies of diagnostische coronair angiografiën. (Kolkailah 2018) |
|
C |
Het is zeer wenselijk vaccinaties subcutaan te geven, in plaats van intramusculair of intradermaal. (Evans 1990, Shrivastava 2013) |
|
B |
Bij grote orthopedische ingrepen geeft continue infusie minder postoperatief bloedverlies, minder Hb-daling en minder noodzaak voor bloedtransfusie. (Park 2017) |
Samenvatting literatuur
Resultaten
Algemeen
Er zijn geen gerandomiseerde studies verricht naar de behandeling van de individuele ingrepen bij patiënten met hemofilie. De data die deze aanbevelingen ondersteunen komen voort uit klinische ervaring, consensus van expert opinions en observationele studies. Er wordt onderscheid gemaakt tussen kleine, middelgrote en grote ingrepen, waarbij met name de minimale behandelduur en de streefspiegels van stollingsfactorcorrectie variëren.
Circumcisie
Een literatuur overzicht en survey in 26 Europese Haemophilia Comprehensive Care settings in 15 verschillende landen over streefwaarden rondom operaties en ingrepen, rapporteerde dat voor circumcisie een factor plasmaconcentratie van 50-60 IE/dL over het algemeen wordt gehanteerd gedurende 2-4 dagen (Hermans 2009).
Naast de correctie van stollingsfactorconcentratie is gebruik van tranexaminezuur en fibrine lijm volgens het Izmier protocol van additionele waarde (Yilmaz 2010). Hierbij werd bij 6% van de patiënten een nabloeding gezien. In een Iraanse studie van 170 patiënten werd vastgesteld dat bloedingscomplicaties niet gerelateerd waren aan leeftijd onder en boven 1 jaar.
Bij een retrospectieve studie in de Mayo Clinics in the USA, werd bij 48 kinderen en jong volwassenen met een mediane leeftijd 15 jaar een circumcisie verricht tussen 2000-2007 (Rodriguez 2010). Twaalf van de 21 patiënten bekend met een stollingsstoornis voor de ingreep, ondergingen de circumcisie met suppletietherapie, 3 hiervan kregen een bloedingscomplicatie (25%). Zevenentwintig patiënten werden pas later gediagnostiseerd met een stollingsstoornis, van deze patiënten kreeg 8 (30%) een bloeding rondom circumcisie. Bloedingscomplicaties waren niet frequenter dan in de populatie zonder stollingsstoornis. Bloedingen traden ook op ondanks adequate suppletietherapie.
In een survey onder kinderarts-hematologen in de USA naar de behandeling rondom neonatale circumcisie in hemofilie patiënten, wordt gerapporteerd dat 14/64 (22%) slechts eenmaal factorconcentraat toediende, 21/64 (32%) 1 tot 2 postoperatieve doses factorconcentraat en 10/64 (16%) tussen 3-5 dagen en 2/64 (3%) 6-10 dagen (Kearney 2015).
Een vergelijkbare low dose factorconcentraat behandeling wordt door Seck et al. gerapporteerd in Senegal, waarbij gedurende 48 uur elke 24 uur een dosis van 30IU/kg factor VIII-concentraat werd gegeven met goed resultaat (Seck 2017). Hiervan presenteerden 5/26 (19.2%) met een bloedingscomplicatie.
Bij langdurige hoge dosis suppletie bestaat het risico op de ontwikkeling van remmers. Een studie in een Egyptisch cohort van 61 patiënten met ernstige hemofilie A zowel “previously untreated” en “minimally treated”, onder de leeftijd van 36 maanden, beschreef de ontwikkeling van remmers in de follow-up periode van 18 maanden (Elalfy 2012). Hiervan ondergingen 25 een circumcisie; 36 niet. Patiënten werden allen on demand behandeld met plasma-derived stollingsfactor VIII-concentraat en hadden geen remmer bij studie inclusie. Twee giften stollingsfactor VIII werden toegediend van 25 IE/kg, de eerste een uur voor de ingreep, de tweede een uur voor verwijdering van het verband. Tevens werd behandeld met een gelatine spons op het wondoppervlak. Remmer bepaling vond na elke 8 exposure days plaats. Er werden geen bloedingen of infecties gezien, behalve in een patiënt die na een gift stollingsfactor VIII (50IE/kg) afdoende was behandeld. Na een mediane duur van 16 exposure days, werd een hoge titer remmers vastgesteld in 7 patiënten, waarvan 3 in de groep met een circumcisie en 4 in de groep die geen circumcisie ondergingen.
Invasieve tandheelkundige ingrepen
Lokale maatregelen zijn gebruikelijk in de tandheelkunde en mondziekten, kaak- en aangezichtschirurgie bij dento-alveolaire chirurgie. Technieken en middelen die worden toegepast (expert opinion) zijn: Alveole-compressie, Tamponeren van de tandkas met fibrinelijm, cellulose spons/gaas, HemCon®, Floseal® (gelatine matrix met trombine) en het gebruik van afdekplaatjes (Bajkan 2020).
De United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO) richtlijn: “Guidance on the Dental Management of Patiënts with Haemophilia and Congenital Bleeding Disorders” beschrijft de huidige aanbevelingen voor het behandelen van patiënten met hemofilie A en B bij tand- of kiesextractie (Anderson 2013). De UKHCDO richtlijn beveelt aan het stollingsfactorgehalte naar minimaal 50-60 IE/dL te brengen voorafgaand aan invasieve tandheelkundige ingrepen. Meestal is een eenmalige dosering voldoende, bij multipele of gecompliceerde extracties kan de substitutie worden herhaald. Dit is mede afhankelijk van eigen stollingsfactor gehalte en moet per patiënt individueel bepaald worden. Bij volwassenen is overhechting van de wond met oplosbare of onoplosbare hechtingen een belangrijke aanvullende maatregel om nabloedingen te voorkomen. Bij kinderen hoeft er niet standaard overhecht te worden bij extractie van melkgebit.
Daarnaast beveelt de UKHCDO richtlijn aan perioperatief antifibrinolytica te geven, zowel bij kinderen als bij volwassenen. Dit kan zowel oraal als intraveneus, waarbij er de dag vóór de ingreep gestart wordt. Bij kinderen is de dosering tranexaminezuur oraal 25-50 mg/kg per 24 uur, verdeeld over drie giften; intraveneus 25 mg/kg in 3xdd. Bij volwassenen is de dosering tranexaminezuur oraal 1g 3-4x per dag. In beide leeftijdsgroepen in principe gedurende zeven dagen voortzetten. Het is wenselijk dit te combineren met tranexaminezuur mondspoeling 5%, eveneens zeven dagen. Orale mondspoeling wordt uitsluitend bij oudere kinderen en volwassenen gegeven, vanwege het risico dat de aanbevolen dosis fors wordt overschreden als de mondspoeling wordt doorgeslikt (UKHCDO en van Galen 2019).
De UKHCDO richtlijn raadt NSAIDs of aspirine voor pijnstilling af. Wel kan paracetamol gegeven worden, eventueel aangevuld door een morfinomimeticum of selectieve COX2 remmer.
Percutane coronaire interventies en coronair angiografieën
De behandeling bij percutane coronaire interventies en coronair angiografieen volgens een lokale Nederlandse richtlijn is mogelijk en veilig (Schutgens 2009, Tuinenburg 2013). In deze prospectieve evaluatie werden negen diagnostische of therapeutische hartkatheterisaties uitgevoerd bij zes hemofilie patiënten (één patiënt met matig ernstige hemofilie B, en vijf patiënten met milde hemofilie A). De factor niveaus lagen tussen de 70 en 130 (mediaan 100) IE/dL tijdens de interventies. Er werden geen trombotische- of bloedingscomplicaties gezien rondom de procedure of tijdens de follow-up.
Een recente Cochrane review toonde aan dat een benadering via de arteria radialis een lager risico op bloedingen heeft dan een benadering via de arteria femoralis (risk ratio 0,54; 95%BI: 0,40-0,74; 20 gerandomiseerde studies in 23.043 niet-hemofilie patiënten (Kolkailah 2018)).
Vaccinaties
Intramusculaire injecties kunnen bij patiënten met hemofilie A of B spierbloedingen veroorzaken (Kulkarni 2001). Er zijn geen gerandomiseerde studies verricht naar de wijze van toediening van vaccinaties bij patiënten met hemofilie A of B en het optreden van bloedingen. Om een spierbloeding ten gevolge van een intramusculaire injectie te voorkomen is er algemene consensus om vaccinaties subcutaan te geven, in plaats van intramusculair of intradermaal (Evans 1990, Srivastava 2013). Als het nodig is een intramusculaire injectie te geven, adviseert de richtlijn van de World Federation of Hemophilia dit kort na een factor gift te doen; een ijspakking op de injectieplaats aan te brengen vijf minuten van te voren; de kleinst beschikbare naald te gebruiken; en gedurende vijf minuten druk op de injectieplaats uit te oefenen (Evans 1990, Kulkarni 2001, Srivastava 2013).
Orthopedische en grote chirurgische ingrepen
Streefspiegels van stollingsfactoren voor orthopedische en grote chirurgische ingrepen zijn afhankelijk van het type ingreep.
De World Federation of Hemophilia richtlijn “Joint replacement surgery in hemophilia” (Wiedel 2010) beveelt aan om preoperatieve stollingsfactor VIII en factor IX waarden te verhogen naar 120 IE/dL. Bij verblijf van meer dan 4 uur op de operatiekamer nogmaals 40 IE/dL stijging van stollingsfactor VIII en IX waarden na te streven. Aansluitend 72 uur postoperatief dalspiegels van 60-80 IE/dL aan te houden en daarna te verlagen naar een dalspiegel van 50 IE/dL tot 14 dagen postoperatief. In week 3-4 dient een dalspiegel van 30-40 IE/dL aangehouden te worden. Aansluitend in week 5 en 6 vlak voor elke fysiotherapiesessie een waarde van 40 IE/dL. Fysiotherapie kan gestart worden op de 1e dag postoperatief, mits ongecompliceerd verlopen en dient minimaal 6 weken gecontinueerd te worden.
In een recentere guideline van de World Federation of Hemophilia “Guidelines for the management of hemophilia” (Srivastava, 2012, Tabel 7.1) worden geen specifieke top- en dalspiegels genoemd voor orthopedische chirurgie, echter voor major surgery wordt een topspiegel van 80-100 IE/dL vermeld, met spiegels van 60-80 IE/dL op dag 1-3; 40-60 IE/dL op dag 4-6 en 30-50 IE/dL op dag 7-14.
De Nederlandse richtlijn “Diagnostiek en behandeling van hemofilie en aanverwante hemostasestoornissen”(2009) beschrijft voor elke chirurgische ingreep een oplaaddosis die voldoende is om de stollingsfactoractiviteit tot 100 IE/dL te doen stijgen, met streefplasmaspiegels van 80-100 IE/dL op dag 1, 50-80 IE/dL op dag 2-5 en 30-50 IE/dL vanaf dag 6, waarbij de suppletieduur bij heupoperaties langer dan drie weken kan duren.
Een Delphi consensus, gepubliceerd in Haemophilia “Target plasma factor levels for personalized treatment in haemophilia: a Delphi consensus statement” (Iorio,2017) bevelen een target dalspiegel aan van 80 IE/dL tijdens hoogrisico chirurgie en grote chirurgie, daarna een dalspiegel van 50-80 IE/dL tot dag 5-7 postoperatief, 30-50 IE/dL vanaf dag 5-7 tot dag 15 en 15-30 IE/dL voor late postchirurgische profylaxe na de 15e dag.
In een Chinese studie bij 11 adolescenten en jongvolwassenen met hemofiliearthropathie werd een artroscopische synovectomie uigevoerd (Zhang 2018). Preoperatieve factorspiegels waren 80-100 IE/dL. De eerste week 60-80 IE/dL en de tweede week 30-40 IE/dL Er werden geen bloedingscomplicaties beschreven in de perioperatie periode.
Bij rugchirurgie bij 5 hemofilie patiënten in Japan, werd een preoperatieve waarde aangehouden van 80 IE/dL tot dag 14 postoperatief en een waarde van ≥ 50 IE/dL daarna. Patiënten verbleven tot dag 28 in het ziekenhuis. Er werden geen bloedingscomplicaties gezien (Kobayashi 2018).
Een Chinese studie bij 19 hemofilie patiënten werden 20 totale heuparthroplastiek operaties uitgevoerd. Hierbij werd opnieuw een spiegel van 100 IE/dL aangehouden op de dag van de operatie en een dalspiegel van 80, 60 en 40 IE/dL op dag 3, 6, en 9 respectievelijk, en vervolgens een dalspiegel van 20-40 IE/dL. Bij een van de patiënten ontwikkelde een haematoom op de rechter dij, maar niet in de heup zelf. Het gerapporteerde intraoperatieve bloedverlies en bloedverlies bij wonddrainage was 715 ml (300-2000 ml) en 713 ml (200-2950 ml) respectievelijk, hetgeen niet meer is dan bij andere hemofilie patiënten en niet-hemofilie patiënten bij een dergelijke operatie in de literatuur (Zhai 2017).
Er is een prospectieve studie gedaan bij 31 patiënten met ernstige hemofilie A, die 42 totale knie arthroplastiek operaties kregen 21 kregen continue infusie, 21 bolusinfusie. Voor de continue infusie werd gestart met een bolusinfusie met een beoogde plasmaspiegel van 100 IE/dL en vervolgens een infusiesnelheid van 4 IU/kg/h aangepast aan kliniek en stollingsfactor VIII levels. De continue infusie werd gedurende 5 dagen gegeven, met een stollingsfactor VIII level van 100 IE/dL gedurende de eerste 3dagen en de dagen daarna van 80 IE/dL. Vanaf dag 6 ging deze groep over op bolusinfusie. De bolusinfusie werd gegeven met een preoperatieve streefspiegel van 100 IE/dL. De eerste 7 dagen werd een dosering van 25 IE/kg elke 8 h gegeven, daarna werd het verminderd naar een 2 maal daagse dosering. In de bolusinjectie groep werd een significant grotere Hb-daling (gemeten in g/dL) gezien 2,26 ± 1,238 versus 1,14 ±0,961 en het bloedvolume in de drain was ook groter 788 ±351 mL versus 574 ±286 mLl op dag 1 postoperatief. 5 van de 21 patiënten in de bolusinjectie groep hadden een bloedtransfusie nodig versus 1 in de continue infusie groep (Park 2017).
Referenties
Bewijskracht literatuur
Level 3 , consistent
Zoeken en selecteren
Circumcisie
Er werd voor deze uitgangsvraag een systematische review verricht vanaf 2010. Zoektermen: “hemophilia”, and “circumcision”. Dit resulteerde in 10 publicaties. Na titel en abstract screening werden 6 publicaties geselecteerd op basis van de volledige tekst, 1 werd geïncludeerd ondanks het ontbreken van volledige tekst, alleen abstract. De overige publicaties werden geëxcludeerd op basis van het ontbreken van relevante informatie over de vraag, of geen abstract beschikbaar, of bij remmers of andere bloedingsziekten.
Invasieve tandheelkundige ingrepen
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van 1 recente evidence based richtlijn, aangevuld door 1 recent (overzichts)artikel.
Percutane coronaire interventies en coronair angiografiën
Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies, geen systematisch literatuuronderzoek verricht. Er werd gebruik gemaakt van een eerder gepubliceerde richtlijn voor de behandeling van ischemische hartziekten in hemofilie patiënten, en een prospectieve evaluatie van die richtlijn. Bij hemofilie patiënten is geen gerandomiseerd onderzoek verricht waarin de radiale benadering met de femorale benadering wordt vergeleken. Er werd voor deze uitgangsvraag, gezien het ontbreken van vergelijkende studies bij hemofilie patiënten, een systematisch literatuuronderzoek verricht onder patiënten in het algemeen. Als eerste werd in de Cochrane Database of Systematic reviews gezocht: ‘pci’ in Title, abstract, keywords (11 hits waarvan 1 relevant protocol wat naar 1 relevante systematische review leidde (Kolkailah, Alreshq et al. 2018). Omdat deze review 20 gerandomiseerde gecontroleerde studies includeert (20.043 patiënten) die bloedingen als uitkomstmaat hebben, is de kans dat een meer recente trial het resultaat van de meta-analyse in relevante mate gaan beïnvloeden verwaarloosbaar klein. Er werd daarom niet verder gezocht naar studies die gepubliceerd zijn na verschijning van deze Cochrane review met zoekdatum oktober 2017.
Vaccinaties
Voor het beleid bij vaccinatie bij hemofilie A en B werd een systematische literatuursearch verricht in met de volgende zoektermen: “vaccination” and “hemophilia” zonder restricties. Dit resulteerde in 90 publicaties, waarvan er drie relevant werden gevonden.
Orthopedische ingrepen
Er werd gebruik gemaakt van de Nederlandse richtlijn, een tweetal richtlijnen van de World Federation of Hemophilia en een Delphi consensus statement. Er werd een additionele literatuursearch verricht vanaf 2012. Zoektermen: “hemophilia”, and “orthopedic surgery”. Dit resulteerde in 256 publicaties. Na titel en abstract screening werden 4 publicaties geselecteerd op basis van de volledige tekst. De overige publicaties werden geëxcludeerd op basis van het ontbreken van relevante informatie over de vraag, of geen abstract beschikbaar, of bij remmers of andere bloedingsziekten.
Zoekverantwoording
De literatuursearch werd verricht in de volgende electronische databases: Pubmed, Cochrane en MEDLINE. Na ontdubbelen zijn de publicaties gecontroleerd op taal (Engels en Nederlands), relevantie (case reports, observationele en retrospectieve studies) en aanwezigheid van volledige tekst.
Overwegingen
In de formule over continue infusie factor VIII en IX wordt de gewenste infusiesnelheid in IE/kg/uur berekend. Er is voor gekozen om geen vaste infusie hoeveelheid aan te bevelen, omdat het aantal eenheden stollingsfactor afhangt van de activiteitswaarde die wordt nagestreefd (zoals aanbevolen in de tabellen 7.1 en 7.2). In de formule wordt de streefwaarde stijging vermenigvuldigd met de klaring. De streefwaarde stijging is het verschil tussen de gewenste streefwaarde minus de eigen basale factor VIII/IX activiteit in IE/dL.
De klaring is een getal dat is afgeleid uit de data afkomstig uit de Nederlandse OPTI-CLOT trial, eenmulticenter Nederlands onderzoek naar perioperatieve substitutie therapie bij hemofilie.
In het model voor factor VIII (Hazendonk 2016):
In model voor factor IX (Preijers 2018):
In de OPTI-CLOT trial data (factor VIII) zijn de volgende klaringen gezien wanneer de samples pre-operatief en net postoperatief worden meegenomen: median (range: 0,024 (0,012 – 0,059) dL/h/kg (leeftijd range 4-76 jaar).
Gezien deze uitkomsten wordt er onderscheid gemaakt in factor VIII en IX en in volwassenen en kinderen, zoals aangegeven in de aanbevelingen.
Tabellen 7.1 en 7.2
De werkgroep komt, conform de aanbevelingen in internationale richtlijnen, tot de aanbevolen streefspiegels en behandelduur zoals aangegeven in Tabel 7.1 en 7.2. Daarnaast neemt de werkgroep algemene maatregelen over in haar aanbevelingen. Voor huidbiopten, kleine dermatologische ingrepen en scopieën met of zonder biopt zijn de aanbevolen streefspiegels en behandelduur gebaseerd op consensus binnen de werkgroep.
Tabel 7.1 Behandeling van hemofilie A en B naar bloedingsrisico van de ingreep
|
Bloedingsrisico |
Initiële top streef spiegel factor VIII/IX (IE/dL) |
Streefdal-spiegel I.T. onderhoud factor VIII/IX (IE/dL) |
Streefspiegel C.I. factor VIII/IX (IE/dL) |
Minimale duur (dagen) |
|
Klein |
50-100 |
– |
– |
eenmalig |
|
Middelgroot |
100 |
> 50 |
50 – 80 |
1 |
|
– |
> 30 |
30 – 50 |
2 – 5 |
|
|
Groot |
100 |
> 50 |
80 – 100 |
1 |
|
– |
> 50 |
50 – 80 |
2 – 6 |
|
|
– |
> 30 |
30 – 50 |
> 7 |
I.T. = intermitterende therapie; C.I. = continue infusie; dag 1 = eerste 24 uur na de ingreep
Tabel 7.2 Behandeling van patiënten met hemofilie A en B bij specifieke ingrepen, naar type ingreep
|
Type ingreep |
Initiële top streef spiegel factor VIII/IX (IE/dL) |
Streefdal-spiegel I.T. onderhoud factor VIII/IX (IE/dL) |
Streefspiegel C.I. factor VIII/IX (IE/dL) |
Gebruikelijke minimale duur (dagen) |
|
Intramusculaire injecties |
50 |
– |
– |
éénmalig |
|
Circumcisie |
100 |
> 50 |
80 – 100 |
1 |
|
– |
> 50 |
50 – 80 |
2 – 4 |
|
|
Ongecompliceerde extractie van 1 tand of kies |
50 |
– |
– |
éénmalig
|
|
Multiple of gecompliceerde tand- of kiesextracties |
50 – 100 |
> 50 |
50 – 80 |
1 |
|
– |
> 30 |
30 – 50 |
2 -5 |
|
|
Percutane Coronaire Interventie |
80 |
> 50 |
50 – 80 |
1 (bij a. femoralis 1 dag extra) |
|
Huidbiopten |
> 50 |
– |
– |
éénmalig |
|
Scopieën zonder biopten |
50 – 100 |
– |
– |
éénmalig |
|
Scopieën met biopten |
100 |
> 50 |
50 – 80 |
1 – 2 |
|
Scopieën met poliepectomie |
100 |
> 50 |
80 – 100 |
1 |
|
– |
> 50 |
50 – 80 |
2+3 |
|
|
– |
> 30 |
30 -50 |
4+5 |
|
|
Kleinere chirurgische ingrepen |
100 |
> 50 |
80 – 100 |
1 |
|
– |
> 50 |
50– 80 |
2+3 |
|
|
– |
> 30 |
30 – 50 |
4+5 |
|
|
Grote chirurgische ingrepen * |
100 |
> 80 |
80 – 100 |
1 |
|
– |
> 50 |
50 – 80 |
2 – 6 |
|
|
– |
> 30 |
30 – 50 |
7 – 14 |
I.T. = intermitterende therapie; C.I. = continue infusie; dag 1 = eerste 24 uur na de ingreep
* gewrichtsvervangende operaties, abdominale chirurgie (incl. darmnaden), cardiothoracale operaties, neurochirurgische operaties
Bij het opstellen van de tabellen 7.1 en 7.2 van deze module is uitgegaan van het standpunt dat er tussen hemofilie A en hemofilie B patiënten geen verschillen in bloedingsneiging bestaan bij een gelijke hoogte van stollingsactiviteit van stollingsfactor VIII, respectievelijk stollingsfactor IX.
De tabellen zijn tot stand gekomen door gebruik te maken van de on line publicatie van de World Federation of Hemophilia “Guidelines for the management of hemophilia” 2012. In die guideline wordt in tabel 7.1 (opgesteld voor landen zonder belangrijke financiële beperkingen) streefspiegels genoemd voor grote chirurgische ingrepen.
Voor grote orthopedische ingrepen werd een literatuur search uitgevoerd. Grote chirurgische en grote orthopedische ingrepen werden vervolgens samengevoegd tot 1 type ingreep.
De genoemde streefspiegels in tabel 7.1 en 7.2 zijn tot stand gekomen op basis van consensus van de werkgroep herziening richtlijn hemofilie, bestaande uit hemofiliebehandelaren voor kinderen en volwassenen; gebruik makend van de ervaringen met de bestaande Nederlandse richtlijn hemofilie uit 2009 en de richtlijnen van de United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO) uit 2010 en 2013.
Hoe is de prenatale diagnostiek en het beleid in de zwangerschap en bij de partus van hemofilie draagsters?
Aanbevelingen m.b.t. de zwangere draagster zelf
Aanbevelingen m.b.t. prenatale diagnostiek
Aanbevelingen betreffende de neonaat met (mogelijk) hemofilie
Onderbouwing
Inleiding
Ongeveer de helft van de vrouwen die draagster is van hemofilie A of B heeft zelf ook een verlaagd factor VIII resp. factor IX gehalte (Veltkamp, 1968). Dit gaat gepaard met een verhoogd risico op bloedingen rondom de bevalling (Chi, 2008; Knol, 2011; Sharathkumar, 2009). Gedurende de zwangerschap stijgt het factor VIII gehalte spontaan twee tot driemaal, factor IX stijgt maximaal anderhalf keer. In de gezonde populatie bedragen ten tijde van de 35-42e week van de zwangerschap de 2,5e en 97,5e percentielen van factor VIII 130 IE/dL en 430 IE/dL en van factor IX 92 IE/dL en 215 IE/dL (Szecsi, 2010). Bij hemofiliedraagsters treedt ook een stijging op, maar deze zal een minder hoge piekwaarde bereiken wanneer de draagster zelf een verlaagd factor VIII resp. factor IX gehalte heeft voor haar zwangerschap. In het kraambed daalt het spontaan gestegen factor VIII bij hemofilie A draagsters bovendien weer vanaf 48 uur postpartum (Huq, 2012; Stoof, 2015).
Conclusies
|
SORT grade |
Conclusies |
|
C |
Voor zwangere hemofilie vrouwen met een verlaagd factor VIII / IX in het derde trimester van de zwangerschap is bevallen in een hemofiliebehandelcentrum sterk gewenst. (expert opinion/consensus) |
|
C |
Een piekspiegel van factor VIII / IX van 150 IE/dL ten tijde van de bevalling is meer fysiologisch dan 100 IE/dL en zou het aantal postpartum bloedingen mogelijk kunnen verminderen. (expert opinion/consensus) |
|
C |
Een minimale duur van de stollingscorrectie met een streefdalspiegel factor VIII / IX ≥ 50 IE/dL van 3 dagen na een spontane vaginale bevalling en 5 dagen na een keizersnede is noodzakelijk. (expert opinion/consensus) |
|
C |
Het gebruik van tranexaminezuur wordt sterk aanbevolen om de kans op zowel een directe als late postpartum bloeding te verminderen. (expert opinion/consensus) |
|
C |
Er is géén contra-indicatie voor het uitvoeren van spinale of epidurale anesthesie mits de factor VIII / IX activiteit ≥ 50 IE/dL is rondom de procedure. (expert opinion/consensus) |
|
C |
Invasieve prenatale diagnostiek wordt sterk aanbevolen bij een mannelijke foetus om hemofilie aan te tonen dan wel uit te sluiten. (expert opinion/consensus) |
|
C |
Bij een foetus met (mogelijk) hemofilie is een atraumatische partus noodzakelijk om de kans op perinatale bloedingen te verminderen. (expert opinion/consensus) |
Samenvatting literatuur
Resultaten
Incidentie postpartumbloedingen bij hemofilie draagsters
In een groot retrospectief Nederlands cohort onderzoek naar 116 bevallingen bij 93 hemofilie draagsters kwam aan het licht dat bijna 30% van de hemofilie draagsters met een factor VIII of IX < 50 IE/dL in het derde trimester een postpartum bloeding kreeg van 500 ml bloedverlies of meer, ondanks profylactische behandeling met streefwaarde van 100 IE/dL ten tijde van de partus. Dit is tweemaal vaker dan in de algemene populatie voorkomt (Stoof, 2015). Van de hemofilie A draagsters met een derde trimester factor VIII waarde tussen 50-100 IE/dL krijgt nog altijd 1 op de 3 vrouwen te maken met een postpartum bloeding (Zwagemaker, 2018), Uit een retrospectief medische status onderzoek verricht in de UK bleek de incidentie van postpartum hemorragie (PPH) na introductie van een richtlijn voor draagsters waarin stollingscorrectie werd geadviseerd bij een derde trimester factor VIII/IX waarde < 50 IE/dL niet significant te dalen (rond 20%), hoewel er een afname werd gezien van de incidentie van ernstige PPH >1 ltr (9% naar 2%) (Chi, 2008). Verder werd door Greer et al. in een oudere retrospectieve studie een ernstige postpartum bloeding gezien bij 2 van 18 hemofilie A draagsters met in totaal 34 zwangerschappen. Deze twee vrouwen hadden een placentarest en een factor VIII waarde 50-100 IE/dL in het derde trimester. Een uterusextirpatie en bloedtransfusies waren nodig om het bloeden te stoppen. Een derde hemofilie A draagster, met een verlaagd eigen factor VIII rond de 15 IE/dL, had een ernstige late postpartum bloeding vier weken postpartum (Greer, 1991). Een Frans universitair ziekenhuis rapporteerde incidentie van 11% primaire en 9% secundaire PPH bij 54 draagsters (32% hemofilie B draagsters) met 101 bevallingen (Sauguet, 2015).
Bij hemofilie B draagsters zijn nog minder data beschikbaar, maar lijkt een postpartum bloeding toch niet zelden voor te komen. In de studie van Stoof et al. had 37% van de hemofilie B draagsters een postpartum bloeding, waaronder drie van de vier vrouwen die preventief factor IX suppletie kregen. In een retrospectief onderzoek naar 50 zwangerschappen van 24 hemofilie A en 8 hemofilie B draagsters in de UK werd 10x een primaire postpartum bloeding gezien, waaronder 3 hemofilie B draagsters (Kadir, 1997). In een ander retrospectief onderzoek werd een postpartumbloeding gerapporteerd bij 4 van 5 draagsters met een factor IX < 20 IE/dL in 6 van 16 zwangerschappen (Yang, 2004). In het bovengenoemde onderzoek van Greer et al. kwam eenmaal een perineaal hematoom waarvoor bloedtransfusie en eenmaal een late postpartumbloeding voor bij 12 zwangerschappen van 5 draagsters met een verlaagd factor IX < 40 IE/dL die profylactisch met fresh frozen plasma waren behandeld (Greer, 1991).
Behandeling rondom de bevalling en prenatale diagnostiek bij hemofilie draagsters
Stollingsfactorcorrectie
Er zijn geen gerandomiseerde of observationele studies verricht naar de meest optimale targetlevels voor factor VIII/IX rondom de bevalling bij hemofilie draagsters. Zoals hierboven beschreven blijkt echter de prevalentie van zowel primaire als late postpartum bloedingen hoog bij hemofilie draagsters, ondanks suppletie van stollingsfactoren met in internationale richtlijnen geadviseerde piek streefspiegel 100 IE/dL ten tijde van de bevalling en dalspiegels > 50 IE/dL gedurende 3-5 dagen in het kraambed (Nichols, 2008; Pavord, 2017).
DDAVP
Er zijn geen gerandomiseerde studies gedaan naar het gebruik van DDAVP ter preventie van of behandeling van bloedingen bij zwangere vrouwen met een aangeboren stollingsstoornis (Karanth, 2015). Er is ervaring opgedaan met het gebruik van DDAVP tijdens de zwangerschap van draagsters van hemofilie A en ook bij de bevalling van vrouwen met de ziekte van Von Willebrand, waarbij een stijging van het factor VIII en von Willebrand factor werd waargenomen en waarbij het middel veilig lijkt te kunnen worden gebruikt (Mannucci, 2005; Trigg, 2012). De meest recente UK richtlijn adviseert mede o.b.v. farmacologische overwegingen om als eerste keus DDAVP 0,3microgram/kg iv. te gebruiken om het factor VIII te corrigeren > 50 IE/dL bij een bekende respons (Pavord, 2017).
Tranexaminezuur
Tranexaminezuur leidt ook bij vrouwen zonder stollingsstoornis tot een significante afname van postpartum bloedingen en is bovendien geassocieerd met een afname van maternale mortaliteit wanneer een postpartumbloeding optreedt (Li, 2017; Shakur, 2017). Na de bevalling daalt het spontaan gestegen factor VIII bij hemofilie A draagsters vanaf 48 uur postpartum (Huq, 2012). Daarnaast neemt de fibrinolyse postpartum toe, door de verminderde synthese van placentair plasminogeen activator inhibitor-2 en een gelijktijdige stijging van tissue plasminogeen activator (Kouides, 2016). In een retrospectief statusonderzoek naar de uitkomst van 62 zwangerschappen bij 33 vrouwen met aangeboren stollingsstoornissen, waaronder 11 hemofilie A draagsters, bleek dat de kans op een ernstige late bloeding in het kraambed significant lager was als tranexaminezuur na ontslag door gebruikt werd, zonder dat dit leidde tot trombose (Hawke, 2016). Er werd geen trombogeen effect gezien van tranexaminezuur in een grote retrospectieve analyse van 256 zwangere vrouwen met een stollingsstoornis, waarvan 168 een keizersnede ondergingen (Lindoff, 1993).
Neuraxiale anesthesie
Choi et al. vonden in de literatuur 107 neuraxiale anesthesie procedures bij 85 hemofilie patiënten, waaronder 7x epidurale anesthesie voor een bevalling. Hierbij trad in géén enkel geval een spinaal hematoom op na stollingscorrectie voorafgaand aan de procedures (Choi, 2009). Chi et al. rapporteerden 11 ongecompliceerde ruggenprikken (epiduraal, spinaal of beide) bij 10 draagsters (9 hemofilie A en 1 hemofilie B) waarvan de hemofilie B draagster vooraf stollingscorrectie kreeg (Chi, 2009). Het risico op ernstige complicaties na neuraxiale anesthesie, zoals een spinaal/epiduraal hematoom, is erg klein, in de orde van 0,85 per 100.000 procedures (95%CI 0-1,8 per 100.000), waarbij de kans iets hoger is na epidurale t.o.v. spinale anesthesie. Vanwege deze lage incidentie is niet te kwantificeren in welke mate dit risico verhoogd is bij hemofilie draagsters, maar waarschijnlijk is het risico klein (Choi, 2009; Harrop-Griffiths, 2013; Moen, 2008). De meest recente UK richtlijn adviseert de factor VIII/IX waarde te corrigeren > 50 IE/dL vooraf insertie en verwijdering van een epidurale catheter en voor spinale anesthesie (Pavord, 2017).
Neonatale uitkomsten
Er zijn geen gerandomiseerde studies verricht met de vraagstelling of een vaginale bevalling vs. primaire keizersnede de veiligste optie is m.b.t. maternale en neonatale uitkomsten bij hemofilie draagsters die zwanger zijn van een (mogelijk) aangedane zoon (Karanth, 2017). De kans op een intracraniële bloeding is sterk verhoogd bij neonaten met hemofilie, waarbij de incidentie rond de 2,5% ligt (Davies, 2016; Kulkarni, 2017; Richards, 2012). Hoewel er een duidelijke associatie bestaat tussen intracraniële bloedingen en een instrumentale vaginale bevalling (forceps/vacuüm extractie), is het verschil in incidentie bij een spontane vaginale partus vs. een keizersnede minder evident. Intracraniële bloedingen zijn ook beschreven bij neonaten met hemofilie na een keizersnede (Davies, 2016; Kulkarni, 2017; Richards, 2012). De meest recente UK richtlijn adviseert om de optie van een primaire keizersnede te bespreken met draagsters die zwanger zijn van een mannelijke neonaat met (mogelijk) ernstige hemofilie, waarbij wordt benadrukt dat de argumenten voor en tegen goed dienen te worden afgewogen (Pavord, 2017).
Referenties
Bewijskracht literatuur
Level 3, consistent
Zoeken en selecteren
Er werd voor deze uitgangsvraag een brede systematische review verricht met de volgende zoektermen: “Inherited Bleeding Disorders”, “Hemophilia”, “Von Willebrand Disease”, “Pregnancy” and “Postpartum Hemorrhage”, zonder restricties. Dit resulteerde in 6788 publicaties waarvan er 5468 overbleven na ontdubbelen. Na titel en abstract screening werden 446 publicaties geselecteerd voor selectie o.b.v. de volledige tekst. Dit resulteerde in 81 relevante publicaties met originele patiënten data t.a.v. beleid rondom de bevalling (59 specifiek over VWD, 19 specifiek over hemofilie en 4 over beiden) en 213 andere relevante publicaties, waaronder reviews en richtlijnen. De overige publicaties werden geëxcludeerd op basis van het ontbreken van originele patiënt data (n=48) of full tekst niet beschikbaar (n=103).
Zoekverantwoording
De literatuursearch werd verricht in de volgende elektronische databases: CENTRAL, Cochrane library, MEDLINE, EMBASE, CINAHL. Na ontdubbelen zijn referentielijsten van geselecteerde artikelen en relevante reviews gecontroleerd op mogelijk relevante studies die gemist konden zijn bij de initiële search. Studies met originele patiëntendata, zowel observationeel (inclusief case series, case reports, cohort studies) als interventie studies (gerandomiseerd/quasi gerandomiseerd) werden geïncludeerd. Artikelen in een andere taal dan Nederlands, Engels of Duits en reviews zijn geëxcludeerd. Eén reviewer heeft titels en abstracts gescreend op mogelijke relevantie. Twee reviewers hebben onafhankelijk van elkaar alle beschikbare volledige teksten van mogelijk relevante artikelen gescreend op de in- en exclusie criteria.
Datum laatste literatuur search: 16-2-2018
Evidence tabellen
Geen.
Overwegingen
Ad aanbeveling 1:
Draagsters van hemofilie hebben bij elke zwangerschap een kans van 1 op de 4 kinderen op een zoon met hemofilie en 1 op de 2 als het kind een zoon is. Bij preconceptionele counseling in een hemofiliebehandelcentrum worden de kans op overerving en de consequenties besproken met de aankomende ouders. Voor ouderparen die willen voorkomen dat ze een zoon met hemofilie krijgen, bestaat de mogelijkheid van prenatale diagnostiek met beëindigen van de zwangerschap (zie figuur 8.1) of pre-implantatie genetisch diagnostiek (PGD) waarbij m.b.v. een IVF procedure niet-aangedane embryo’s kunnen worden geselecteerd en teruggeplaatst. Deze preconceptionele counseling dient bij voorkeur plaats te vinden door de klinisch geneticus, die dan tevens dient te checken of de oorzakelijke mutatie bekend is of DNA-onderzoek dient te starten. Beleid rondom de bevalling bij hemofilie draagsters dient bij voorkeur bepaald te worden in een vast team tenminste bestaande uit een internist-hemofiliebehandelaar, gynaecoloog-perinatoloog, anesthesist, kinderarts-hematoloog en klinisch geneticus in een hemofiliebehandelcentrum.
Ad aanbeveling 2:
Het advies om de bevalling in een hemofiliebehandelcentrum te laten plaatsvinden is gebaseerd op de aanwezige expertise voor moeder en kind, vergoeding van stollingsfactoren en 24u beschikbaarheid van laboratoriumfaciliteiten om de factor VIII/IX activiteit te monitoren rondom de bevalling. Ruim voor de uitgerekende datum dient een partusplan te zijn opgesteld i.o.m. de hemofilie behandelaar, gynaecoloog, anesthesioloog en kinderarts. Het advies is om bij uiterlijk 24 weken zwangerschap een voorlopig partusplan klaar te hebben en dit zo nodig bij te stellen o.b.v. bereikte stollingswaarden in het derde trimester. Dit plan wordt gedocumenteerd en besproken. In dit partusplan staat minimaal beschreven wat de diagnose is, het stollingsbeleid afhankelijk van de modus partus, het beleid ten aanzien van het kind, het beleid voor de anesthesioloog en eventuele noodzakelijke stollingscorrectie in het kraambed.
Voor hemofiliedraagsters waarbij in het derde trimester van de zwangerschap het factor VIII resp. factor IX gehalte is gestegen tot > 80 IE/dL en waarbij de vrouw zwanger is van een meisje of niet aangedane jongen, hangt de locatie van de bevalling af van het individuele bloedingsfenotype van de vrouw en dient de locatie van de bevalling in het multidisciplinaire overleg te worden vastgesteld. Deze vrouwen mogen bij geen of zeer minimale, persoonlijke bloedingsanamnese eventueel ook in de eerste lijn bevallen.
Ad aanbeveling 3:
Het feit dat hemofiliedraagsters met een verlaagd factor VIII of IX niet dezelfde piekwaardes bereiken rondom de bevalling als vrouwen met een normaal factor VIII of IX vormt een mogelijke verklaring voor de verhoogde incidentie van postpartum bloedingen in deze categorie vrouwen. Een ernstige postpartum bloeding, vanaf 1000 ml bloedverlies in de eerste 24uur na de bevalling, is sterk geassocieerd met een verhoogde kans op maternale sterfte (Dahlke et al. 2015). Daarom is in de huidige richtlijn besloten om een hogere, meer fysiologische, piekspiegel van factor VIII en IX aan te houden ten tijde van de bevalling van 150 IE/dL.
De intensiteit van de stollingsmedicatie rondom de bevalling is afhankelijk van het stolfactorpercentage in het derde trimester van de zwangerschap, waarbij het doseringsadvies uit de onderstaande tabel 8.1 als leidraad kan worden gebruikt. Er is bewust voor gekozen om DDAVP pas te gebruiken ná het afklemmen van de navelstreng, i.v.m. de beperkte ervaring tijdens de zwangerschap en mogelijke bijwerkingen voor de neonaat.
Tabel 8.1: Peripartum stollingscorrectie bij zwangere draagsters van hemofilie A en B
|
Stolfactorgehalte |
In partu¥ |
Na afklemmen navelstreng¥ |
Bij fluxus ≥ 500ml*¥ |
Kraambed¥ |
|
Hemofilie A draagster |
|
|
|
|
|
FVIII < 50 IE/dL preconceptioneel én 3e trim FVIII ≥ 80 IE/dL |
– |
Overweeg TXA |
TXA indien nog niet gegeven en DDAVP
|
TXA on demand
|
|
3e trim FVIII < 80 IE/dL |
FVIII-concentraat |
TXA |
Overweeg extra FVIII-concentraat of DDAVP |
TXA én FVIII of DDAVP indien FVIII < 50 IE/d zakt |
|
Hemofilie B draagster |
|
|
|
|
|
FIX < 50 IE/dL preconceptioneel én 3e trim FIX ≥ 80 IE/dL |
– |
Overweeg TXA |
TXA indien nog niet gegeven |
TXA on demand
|
|
3e trim FIX < 80 IE/dL |
FIX-concentraat |
TXA |
Overweeg extra FIX-concentraat |
TXA én FIX indien FIX < 50 IE/d zakt |
TXA= Tranexaminezuur; FVIII= Factor VIII activiteit; FIX= Factor IX activiteit; DDAVP= desmopressine (Minrin®/Octostim®)
¥ Zie tekst hieronder voor Streefwaarden en Dosering
* daarnaast behandeling volgens het lokale fluxus protocol
Streefwaarden
Doseringen
NB Er is geen contra-indicatie voor profylactisch LMWH in het kraambed zolang de stolling volledig gecorrigeerd/genormaliseerd is. Het gebruik van DDAVP, tranexaminezuur en/of stollingsfactor vormt géén contra-indicatie voor het geven van borstvoeding (https://www.lareb.nl/teratologie-nl/borstvoeding/).
Ad aanbeveling 4:
In deze richtlijn wordt 1000 mg tranexaminezuur intraveneus aanbevolen ten tijde van de bevalling, na het afklemmen van de navelstreng, omdat dit ook bij vrouwen zonder stollingsstoornis leidt tot een verminderd risico op een postpartumbloeding en op een verminderd risico op maternale sterfte als een postpartumbloeding optreedt. Het advies om tranexaminezuur in het kraambed te continueren bij draagsters met een derde trimester factor VIII/IX waarde < 50IE/dL is gebaseerd op het feit dat het spontaan gestegen factor VIII bij hemofilie A draagsters vanaf 48 uur postpartum weer daalt en op literatuurgegevens dat het aantal late post partum bloedingen verminderde wanneer tranexaminezuur langer door gebruikt werd (Hawke, 2016). Het is onbekend hoe lang Tranexaminezuur dient te worden voorgezet na de bevalling. In de studie van Hawke et al, varieerde de behandelduur van 5 dagen tot 6 weken postpartum (mediaan 3 weken, n=36). Het lijkt raadzaam om de behandeling met Tranexaminezuur in elk geval te continueren zolang de noodzakelijke frequentie van het verschonen van kraamverbanden nog hoog is (bijvoorbeeld vaker dan elke 3 uur). Het gebruik van Tranexaminezuur vormt géén contra-indicatie voor het geven van borstvoeding. Het middel gaat in kleine hoeveelheid wel over in de moedermelk, maar hiervan zijn geen schadelijke effecten bekend (https://www.lareb.nl/teratologie-nl/borstvoeding/).
Ad aanbeveling 5:
Het risico op ernstige complicaties na neuraxiale anesthesie, zoals een spinaal/epiduraal hematoom wordt bij hemofilie draagsters na spontane normalisatie of o.b.v. stollingsfactorsuppletie de literatuur als zodanig laag ingeschat dat dit in elk geval niet opweegt tegen het toegenomen risico van algehele anesthesie bij een keizersnede. Ook voor het ontraden van epidurale anesthesie t.b.v. pijnstilling tijdens de bevalling zijn de bloedingsrisico’s te laag. Wel is aan te raden, met het oog op de spaarzame literatuurgegevens voor deze categorie patiënten, om ook alternatieve mogelijkheden voor pijnstilling te bepreken. In overeenstemming met internationale richtlijnen is in de huidige richtlijn besloten dat er géén contra-indicatie bestaat voor neuraxiale anesthesie na normalisatie of correctie van de factor VIII/IX activiteit ≥ 50 IE/dL bij zwangere draagsters rondom de bevalling.
LET OP deze streefwaarden gelden óók bij het verwijderen van een eventueel epiduraal katheter. In elk geval zouden alle zwangere draagsters de mogelijkheid moeten krijgen om de voor- en nadelen van de verschillende opties voor pijnstilling tijdens de bevalling vooraf te bespreken met een ervaren anesthesioloog.
Ad aanbeveling 6:
Zwangeren die (mogelijk) bekend zijn met dragerschap van hemofilie horen gezien te worden vroeg in het eerste trimester in een hemofiliebehandelcentrum. Het advies is hen tevens te verwijzen naar een de klinisch geneticus. De klinisch geneticus kan aan de hand van de familieanamnese nazoeken of de oorzakelijke mutatie bij patiënte of in de familie reeds bekend is ofwel genetisch onderzoek verrichten (bij voorkeur echter preconceptioneel, zie Ad 1). Indien dragerschap vast staat is verwijzing geïndiceerd naar een gynaecoloog-perinatoloog met ervaring in het verrichten van invasieve prenatale diagnostiek en naar de internist-hemofiliebehandelaar.
Wanneer invasieve diagnostiek plaatsvindt dient hieraan voorafgaand een geslachtsbepaling van de foetus plaats te vinden. Vanaf de 8e week van de zwangerschap kan met behulp van een Y-PCR in het bloed van de moeder worden bepaald of er sprake is van een mannelijke foetus (zie Figuur 8.1). Als er sprake is van een mannelijke foetus, dan is er een indicatie voor invasieve prenatale diagnostiek. Bij een vrouwelijke foetus is geen indicatie voor invasieve prenatale diagnostiek. Het tijdstip van eventuele invasieve prenatale diagnostiek hangt samen met de keuze van de zwangere en haar partner. Als het paar geen invasieve diagnostiek wenst of wanneer invasieve diagnostiek pas rond 32-34 weken wordt verricht, kan door middel van geavanceerd ultrageluidsonderzoek (echo) rond 20 weken het geslacht van de foetus worden bepaald ten behoeve van het partusbeleid.
Indien het paar kiest voor beëindiging van de zwangerschap als de mannelijke foetus hemofilie heeft, dan kan met een vlokkentest in het eerste trimester of een vruchtwaterpunctie in het tweede/begin derde trimester worden bepaald of de foetus hemofilie heeft. Invasieve diagnostiek vóór 24 weken amenorroe wordt enkel verricht als de zwangere de intentie heeft een zwangerschap af te breken bij een aangedane zoon. De kans op een miskraam door invasieve prenatale diagnostiek in het eerste (vlokkentest) en tweede trimester (vruchtwaterpunctie) zijn respectievelijk 0,2% en 0,1%.. (zie NOTA invasieve prenatale diagnostiek NVOG: https://www.nvog.nl/kwaliteitsdocumenten/notas/).
Indien het paar niet kiest voor beëindiging van de zwangerschap dan is rond 32-34 weken zwangerschap een vruchtwaterpunctie een optie bij een mannelijke foetus. Het doel van de vruchtwaterpunctie is in die situatie aanpassen van het beleid durante partu indien de foetus hemofilie heeft. Aangezien dan een atraumatische partus wordt nagestreefd. Omdat het advies ‘atraumatische partus’ bij een zoon met hemofilie er in de praktijk toe leidt dat sneller wordt overgegaan tot een keizersnede met daarbij behorende hogere maternale risico’s, is het belangrijk om in het geval van een mannelijke foetus bij een hemofilie draagster de mogelijkheid van prenatale diagnostiek expliciet ten behoeve van het obstetrische beleid met het paar te bespreken. De maternale risico’s van een keizersnede zijn niet alleen geldend voor de index zwangerschap maar ook voor de zwangerschappen daarna. Er is geen of zeer minimale verhoogde kans op een vroeggeboorte of foetale complicatie bij een vruchtwaterpunctie in het derde trimester.
Het advies om vooraf aan een eventuele vlokkentest / vruchtwaterpunctie dient de factor VIII/IX waarde eenmalig te corrigeren ≥ 50 IE/dL is mede gebaseerd op het advies in de meest recente UK richtlijn.(Pavord et al. 2017)
Als er sprake is van hemofilie bij de foetus dan bestaat er een indicatie voor een bevalling in een ziekenhuis met een hemofiliebehandelcentrum i.v.m. het risico op een intracraniële bloeding bij de geboorte (zie ook Ad 7). Als niet bekend is of de foetus is aangedaan met hemofilie, is er ook een indicatie voor een bevalling in een ziekenhuis met een hemofiliebehandelcentrum.
Figuur 8.1: stroomschema prenatale diagnostiek bij hemofiliedraagsters
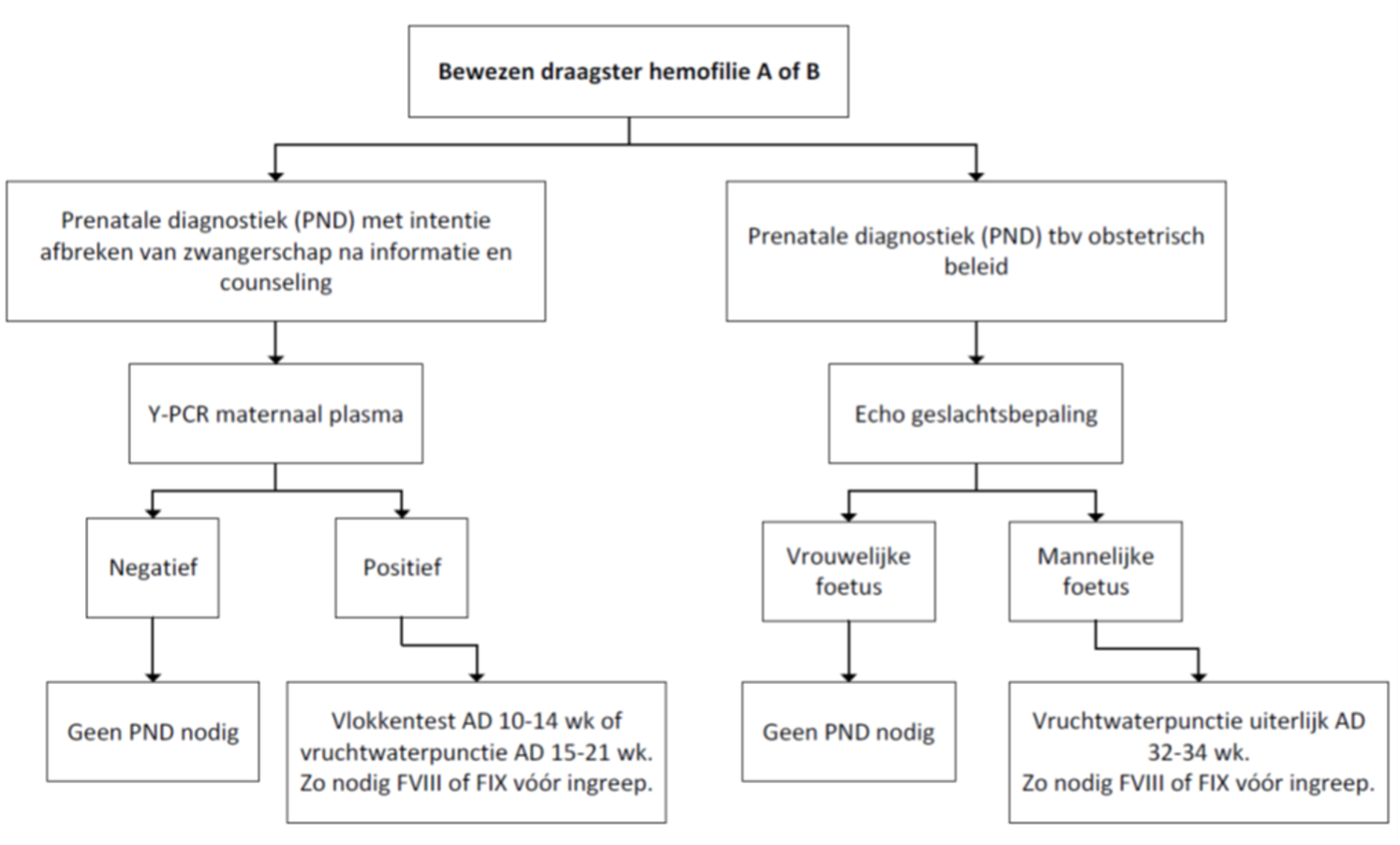
NB Vooraf aan een eventuele vlokkentest / vruchtwaterpunctie dient zo nodig de factor VIII/IX waarde gecorrigeerd te worden met eenmalig een beoogde piekspiegel ≥ 50 IE/dL. PND=prenatale diagnostiek.
Ad aanbeveling 7:
Internationaal is het debat of er een indicatie bestaat voor een primaire keizersnede bij een neonaat met ernstige hemofilie niet beslecht (Madan, 2010). Echter omdat een keizersnede het risico op een intracraniële bloeding bij de neonaat niet uitsluit en omdat een keizersnede meer bloedingsrisico’s met zich meebrengt voor hemofilie draagsters t.o.v. een vaginale partus en een hogere kans geeft op complicaties bij een volgende zwangerschap, is het advies in deze richtlijn om een keizersnede alléén te verrichten op obstetrische indicaties. Het is wel evident dat een instrumentele vaginale partus het risico op een intracraniële bloeding bij de neonaat met hemofilie verhoogt. Daarom dient de bevalling van een mannelijke neonaat met (mogelijk) hemofilie zo atraumatisch mogelijk te verlopen. In algemene zin wordt het risico voor de vrouwelijke foetus indien draagster als zeer minimaal ingeschat. Er zijn zeker uitzonderingen mogelijk met in zeldzame gevallen vrij lage factor spiegels in een draagster, maar daar wordt voor het peripartum beleid geen rekening mee gehouden. Bij een ‘atraumatische partus’ zijn het plaatsen van een schedelelektrode, het doen van microbloedonderzoek, een vaginale stuitbevalling, vaginale kunstverlossing (vacuüm- en forcipale extractie) gecontra-indiceerd. Ook is een evt. versie bij een stuitligging gecontra-indiceerd bij een neonaat met (mogelijk) hemofilie. Daarnaast zijn intramusculaire injecties bij de neonaat gecontra-indiceerd. In noodsituaties waarbij conversie naar een sectio meer risico’s oplevert dan een kunstverlossing, heeft een forcipale extractie de voorkeur boven een vacuümextractie. Bij een instrumentele bevalling dient altijd de kinderarts geconsulteerd te worden om een intracraniële bloeding uit te sluiten en zo nodig met spoed bloedstollingsdiagnostiek in te zetten.
De behandeling van hemofilie wordt vaak gecompliceerd door de ontwikkeling van neutraliserende antistoffen tegen de toegediende stollingsfactor. In deze uitgangsvraag worden in een aantal subvragen aanbevelingen geformuleerd hoe de kans op de de vorming van een remmer zo laag mogelijk te houden, welke behandeling kan worden ingezet om een remmer te doen verdwijnen, en hoe bloedingen bij een patiënt met een remmer voorkómen en behandeld kunnen worden.
Hoe kan de kans op vorming van een remmer zo laag mogelijk worden gemaakt?
Aanbevelingen
Onderbouwing
Inleiding
Bij de behandeling van hemofilie patiënten met stollingsfactorconcentraat kunnen remmende antistoffen ontstaan. Als gevolg hiervan worden de toegediende stollingsfactorconcentraten niet of veel minder werkzaam. Ook neemt de ernst van de bloedingen vaak toe. Bij patiënten met milde en matig ernstige hemofilie kan het endogene FVIII worden afgebroken waardoor zij een vergelijkbaar bloedingsfenotype krijgen als patiënten met ernstige hemofilie.
De definitie van een remmer of inhibitor is een polyclonaal IgG-antilichaam met hoge affiniteit, gericht tegen factor VIII of factor IX. Het antilichaam versnelt de klaring van de toegediende stolfactor waardoor de functionele activiteit van factor VIII of factor IX geheel of gedeeltelijk vermindert; bij een hoge titer remmer is geen opbrengst van toegediend factor VIII of factor IX meetbaar.
Bij hemofilie A zijn de meeste antistoffen gericht tegen het A2-domein van de zware keten of het A3- en C2-domein van de lichte keten van het factor-VIII-molecuul. Bij de meeste patiënten zijn de antistoffen tegen meer dan één epitoop gericht (Pratt, 2016). Bij hemofilie B gaat het optreden van remmers frequent gepaard met een allergische reactie of anafylaxie tegen toegediend factor IX waarbij vaak een nefrotisch syndroom optreedt.
Vooral als de remmer in een hoge titer aanwezig is, belemmert deze de behandelmogelijkheden, waardoor ernstige morbiditeit en mortaliteit ontstaan. Hiermee is het ontstaan van remmers momenteel de ernstigste complicatie van hemofilie.
Conclusies
|
SORT |
Conclusie |
|
A |
Piek behandelingen met factor VIII (dagdosis minstens 50 E/kg gedurende minstens 5 opeenvolgende dagen) ten tijde van de eerste behandeling met FVIII zijn geassocieerd met een verhoogd risico op remmer vorming. (Gouw, 2007; Marcucci, 2015) |
|
B |
Beschikbare literatuur geeft geen basis voor definitief advies ten aanzien van de keuze voor een recombinant of uit plasma vervaardigd factor VIII-concentraat voor de initiële behandeling van ernstige hemofilie A. |
|
A |
In meerdere grote cohortstudies over de initiële behandeling van patiënten met ernstige hemofilie A wordt gerapporteerd dat bij gebruik van Kogenate®/Helixate® een verhoogde incidentie van remmers optreedt. (Gouw, 2013; Calvez, 2014; Björkman, 2013; Fischer, 2015) |
|
B |
Een aantal cohort studies suggereert dat het vroeg starten van profylaxe de remmer incidentie verlaagt. (Gouw, 2007; Gouw, 2013; Kurnik, 2010) |
Samenvatting literatuur
Resultaten
Epidemiologie van remmende antistoffen
Remmende antistoffen tegen factor VIII (factor VIII remmers) komen voor bij ±33% van de kinderen met ernstige hemofilie A en remmende antistoffen tegen factor IX (factor IX remmers) bij 5-10% van de kinderen met ernstige hemofilie B. Remmende antistoffen tegen factor VIII en IX treden vooral op in de eerste 50 behandeldagen (exposure days : EDs) met een mediaan van 14 EDs (Berg van den 2019). Recent heeft de PedNet groep gerapporteerd dat in 1038 previously untreated patients (PUPs) met ernstige hemofilie A 99,3% van alle remmers ontstond in de eerste 75 EDs, wat op een mediane leeftijd van 2,3 jaar (interquartile range 1,7-2,8 jaar) al bereikt werd (van den Berg et al, 2019). Bij patiënten met matig ernstige en milde hemofilie A is de incidentie van remmer ontwikkeling veel lager. De INSIGHT onderzoeksgroep heeft een cumulatieve incidentie beschreven van 6,7% binnen de eerste 50 EDs en 13,3% binnen de eerste100 EDs en deze stijgt nog door met toename van het aantal behandeldagen tot rond de 17% (Eckhardt, 2015) .
Met de introductie van recombinante stollingsfactorconcentraten is de behandeling met stollingsfactorconcentraten geïntensiveerd (Nijdam, 2015) en werden frequenter remmers gediagnosticeerd. Men veronderstelde, dat dit eerder optrad bij een behandeling met recombinant factor VIII. Een recente analyse van 926 PUPS met ernstige hemofilie A liet zien, dat sinds 1990 het aantal hoge titer remmers (ofwel high responders) met een remmer titer van ≥5 BU stabiel rond de 20% is gebleven en dat het aantal gediagnosticeerde patiënten met een lage titer remmers (low responders, max remmer titer < 5 BU) is gestegen van 3,1% naar 10,5 % (Berg van den, 2015).
Etiologie
Over de etiologie van remmer ontwikkeling is veel onderzoek gedaan. Voor een overzicht van de immunologische aspecten verwijzen we naar een recente review. (Schep, 2018).
Er is nog geen duidelijk inzicht in het ontstaansmechanisme van remmers. Wel is een aantal risicofactoren voor remmer vorming geïdentificeerd. Deze worden verdeeld in patiënt-gerelateerde (endogene) en behandelings-gerelateerde (exogene) risicofactoren.
Endogene risicofactoren voor remmer ontwikkeling
Genetische risicofactoren voor remmer ontwikkeling bij patiënten met hemofilie zijn het beste onderzocht. Voor patiënten met ernstige hemofilie A is het bekend dat het risico op een remmer het hoogste is bij patiënten met een factor VIII nulmutatie, waardoor er in het geheel geen factor VIII wordt gemaakt. Dit betreft grote gen deleties, nonsense-mutaties en intron-22-inversies. Tevens is duidelijk dat patiënten met missense mutaties een relatief laag risico hebben op remmers (Oldenburg, 1997)). Daarnaast zijn er aanwijzingen dat variaties in het immuun profiel een rol spelen (Pavlova, 2009; Astermark 2006). Al is hierover nog geen consensus. De bevinding dat polymorfismen in het IL10 gen geassocieerd zijn met remmer ontwikkeling werd niet bevestigd in een Italiaans patiënten cohort (Bafunno, 2009). In een cohort van 823 PUPs met ernstige hemofilie A werden de onafhankelijke bijdragen van verschillende risicofactoren gekwantificeerd. Hierbij waren een positieve familieanamnese voor remmers (OR 2.1; 95% CI 1,3-3,7) en het type factor VIII genmutatie significante risicofactoren. Vergeleken met een missense mutatie hadden patiënten met een nulmutatie een OR van 8,1 (95% CI 3,3-18,9) en die met een andere mutatie een OR van 4,2 (95% CI 1,8-10,8) (Hashemi, 2015). Bij deze laatste studie werd het onafhankelijke effect van het immuunprofiel niet meegenomen.
Exogene risicofactoren voor remmer ontwikkeling
Behandelingsgerelateerde factoren kunnen ook een belangrijke rol spelen bij de ontwikkeling van remmers (Marcucci, 2015; Gouw, 2007, 2013). Een eerste intensieve behandeling met factor VIII (meer dan vijf opeenvolgende expositiedagen met een minimale dosis van 50 E/kg/dag) is geassocieerd met 50% kans op remmer ontwikkeling bij patiënten met ernstige hemofilie A (Gouw, 2007; Marcucci, 2015). In een meta-analyse van individuele data van 761 PUPs met ernstige hemofilie A vonden Marcucci et al. zelfs dat intensieve behandeling bij eerste blootstelling aan factor VIII de belangrijkst onafhankelijke voorspeller was van remmer ontwikkeling (Marcucci, 2015). Een vroege start met behandeling met factor VIII is op zichzelf geen risicofactor voor remmer ontwikkeling gebleken (Hashemi, 2015; Marcucci, 2015).
Ook bij patiënten met milde hemofilie A is aangetoond dat intensieve behandeling, grote bloedingen en chirurgie risicofactoren voor remmer ontwikkeling zijn (Sharathkumar, 2003, Eckhardt, 2009).
Daarnaast lijkt vroege profylactische behandeling een beschermend effect te hebben op remmer ontwikkeling. In diverse onderzoeken, waaronder het CANAL-onderzoek, werd profylactische behandeling geassocieerd met een verlaagd risico van remmer ontwikkeling (Gouw, 2007). Maar bij herhaald onderzoek in het PedNet cohort bleek het beschermende effect van profylaxe pas na 20 EDs op te treden en vooral bij lage titer remmers (Gouw, 2013). Daarna publiceerden Duitse collega’s dat remmers werden voorkomen met zeer vroege profylaxe in een lage dosis (1x per 2wk 250 E) (Kurnik, 2010). Echter, in een prospectieve studie met deze behandelstrategie werd een percentage van 42% remmers gezien, waarna de studie voortijdig werd gestaakt (Auerswald, 2015).
Naar de effecten van de keuze van factor VIII-concentraat op remmer ontwikkeling is veel onderzoek gedaan. Een eerste studie was gebaseerd op de PedNet data, die liet zien dat Kogenate®/Helixate® geassocieerd was met een 60% hogere kans op remmers (Gouw, 2013). Vervolgens werd in een aantal andere cohort studies steeds een trend gezien ten nadele van Kogenate®/Helixate®, waarbij de statistische significantie niet in elke studie werd bereikt. De remmer ontwikkeling op Kogenate®/Helixate® ten opzichte van de remmer ontwikkeling op Advate® was in het Franse cohort verhoogd met een adjusted Hazard Ratio van 1,55 (95% CI 0,97-2,49) (Calvez, 2014); in het Engelse cohort (Björkman, 2013) een adjusted Hazard Ratio van 2,14 (95% CI 1,12-4,10) en in het Europese EUHASS cohort(Fischer 2016) het minste verhoogd met een relatief risico van 1,09 (22 vs. 24% met 95% CI 0,58-2,04). Op grond van deze data wordt aangeraden PUPs met ernstige hemofilie A geen Kogenate®/Helixate® meer voor te schrijven.
Al langer wordt gespeculeerd over de invloed van het type factor VIII-concentraat (plasma dan wel recombinant) op de kans op remmer ontwikkeling. Er zijn verschillende publicaties, die suggereren dat patiënten behandeld met een uit plasma vervaardigd factor VIII-concentraat minder remmers ontwikkelen. Omdat de intensiteit van de behandeling een belangrijke risicofactor voor remmer ontwikkeling is en er voor 2000 veel meer uit plasma vervaardigd factor VIII-concentraten werden gebruikt, werden alleen de publicaties van (internationale) cohorten en een meta-analyse sinds 2000 in overweging genomen (zie tabel 9.1.1).
Deze tabel laat duidelijk zien dat de controverse rondom het mogelijk beschermende effect van uit plasma vervaardigde factor VIII-concentraten nog niet voorbij is. In de gerandomiseerde SIPPET studie werd een significant beschermend effect van Von Willebrand Factor bevattend uit plasma vervaardig factor VIII gevonden (Peyvandi, 2016). Deze resultaten worden niet bevestigd in grote cohortstudies en een meta-analyse. Sommigen zijn van mening dat de SIPPET studie (Peyvandi, 2016) overtuigend bewijs levert dat recombinant factor VIII-concentraten geassocieerd zijn met een hogere remmer incidentie, omdat het studie design het meest sterke bewijs levert. Anderen wijzen op de hogere overall remmer incidentie, de heterogeniteit van de gebruikte concentraten en de lagere behandelingsintensiteit gebruikt in de meerderheid van de deelnemende centra. Feit is wel dat pooling van al deze data een licht verhoogd remmer-risico suggereert. Er zijn echter data van individuele concentraten nodig om een keuze te maken in de kliniek.
Tabel 9.1.1 Remmer incidenties bij uit plasma vervaardigd factor VIII en recombinant factor VIII
|
|
Studie opzet |
pdFVIII |
rFVIII |
Remmers Totaal (%) |
Ratio rFVIII/pdFVIII RR (95% CI) |
|
Franchini, 2012 |
Meta analyse |
83/360 |
115/415 |
25,5 |
1,15 (0,95-1,38) |
|
Collins, 2015 (UKHCDO) |
Cohort UK |
0 |
95/338 |
28,1 |
– |
|
Fischer, 2016 (EUHASS) |
Internationaal register |
5/20 |
37/178 |
21,2 |
1,09 (0,58-2,04) |
|
Gouw, 2013 (PedNet) |
Internationaal register |
29/88 |
145/476 |
31,2 |
0,96 (0,62-1,49) |
|
Calvez, 2014 (FranceCoag) (pd FVIII gegevens van presentatie EAHAD in 2016) |
Cohort FR |
23/110 |
114/303 |
33,2 |
1,80 (1,22-2,66) |
|
Peyvandi, 2016 (SIPPET) |
RCT |
29/125 |
47/126 |
35,4 |
1.87 ( 1.17-2.96) |
|
|
|
|
|
|
|
|
Totaal alle remmers |
|
169/703 (24,0%) |
553/1836 (30,1%) |
28,4 |
1,25 (1,08-1,45) |
Referenties
Bewijskracht literatuur
De bewijskracht uit de literatuur is laag. Er is slechts één direct vergelijkende, gerandomiseerde studie gedaan naar de invloed van gebruik van uit plasma vervaardigd factor VIII-concentraat versus gebruik van recombinant factor VIII-concentraat op de ontwikkeling van remmers bij PUPs. Verder is er alleen sprake van cohortonderzoeken, case histories en consensus op basis van expert opinion. Voor factor IX-concentraten ontbreken studies geheel.
Level 2, inconsistent.
Zoeken en selecteren
Er werd geen systematische search gedaan. Er werd gebruik gemaakt van bekende richtlijnen en de daarin vermelde relevante referenties en bij de auteurs bekende relevante publicaties.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Er is een evidence tabel (tabel 9.1.1) over de mogelijke invloed van gebruik van uit plasma vervaardigd factor VIII-concentraat versus gebruik van recombinant factor VIII-concentraat op de ontwikkeling van remmers bij PUPs.
Overwegingen
Ondanks dat het optreden van remmers de meest frequente complicatie is bij de behandeling van hemofilie patiënten en de ernstige gevolgen die een remmer heeft op de ziektelast en het risico van overlijden, zijn er geen goede studies beschikbaar om goede behandelkeuzes te kunnen maken. Er is slechts één gerandomiseerde studie gedaan naar de mogelijke invloed van behandeling met uit plasma vervaardigd factor VIII-concentraat in vergelijking met recombinant factor VIII-concentraat op de frequentie van het optreden van remmers bij PUPs: de SIPPET studie. Er is internationaal echter veel discussie over de resultaten van deze SIPPET studie, zodat de controverse over deze keuze voortduurt. Bovendien zijn de in de SIPPET studie gebruikte uit plasma vervaardigde concentraten in Nederland niet beschikbaar, wat de keuze over het te voeren beleid ook bemoeilijkt. Daarom wordt aanbevolen de keuze met de ouders te bespreken en tot een gezamenlijke afweging te komen.
Uit cohort studies komen redelijk sterke aanwijzingen naar voren dat een intensieve eerste behandeling met factor VIII een hoger risico geeft op remmervorming. Hieronder wordt verstaan een piekbehandeling gedurende 5 opeenvolgende dagen met minstens 50 E/kg/dag.
Welke behandeling kan worden ingezet om een remmer te doen verdwijnen?
Aanbevelingen
Onderbouwing
Inleiding
Om bloedingen te voorkomen, is bij ernstige hemofilie levenslange profylaxe geïndiceerd. De aanwezigheid van een remmer interfereert met deze behandeling. Daarom is het nodig om zo snel mogelijk tolerantie te bereiken voor factor VIII dan wel factor IX bij alle patiënten met ernstige hemofilie A dan wel B met een remmer.
Conclusies
|
SORT Grade |
Conclusie |
|
A |
Kinderen met ernstige hemofilie A en een remmer moeten zo vroeg mogelijk immuuntolerantie inductie (ITI) therapie krijgen omdat remmer activiteit interfereert met de effectiviteit van profylaxe en daardoor interfereert met de preventie van gewrichtsschade. (Nakar, 2015; Carcao, 2019; Mancuso, 2017; Collins, 2017) |
|
C |
ITI wordt meestal gestart met hetzelfde factor VIII-concentraat waarop de remmer is ontwikkeld (veelal recombinant factor VIII). Er is tot nu toe onvoldoende bewijs dat een uit plasma vervaardigd factor VIII-concentraat met een hoog VWF gehalte of recombinant factor VIII-Fc een superieur resultaat geeft. (Mannucci, 2012; Kreuz, 2016; Oldenburg, 2014; Santagostino, 2019; Rangarajan, 2014; van Velzen, 2014; Carcao, 2018) |
|
B |
Bij lage titer remmers (titer steeds < 5 BU) is gewone profylaxe ± 25-50 E/kg 3x per week of om de dag waarschijnlijk voldoende (Hay 2006; Valentino 2015; Ter Avest 2010; Carcao 2019). Hoge dosis ITI is gecontra-indiceerd omdat dit de kans op progressie naar een hoge titer remmer 4 x verhoogt. (Mancuso, 2017) |
|
B |
Bij patiënten met remmers en een piek titer tussen de 5 en 40 BU wordt geadviseerd lage dosis ITI toe te passen, bij piek titers tussen de 40 en 200 BU is er geen voorkeur voor hoge of lage dosis ITI. (Ter Avest, 2010; Hay, 2012) |
|
C |
Bij patiënten met een piek titer tussen 5 en 200 BU met bloedingen moet overwogen worden om de frequentie van ITI (eventueel in combinatie met dosis) te verhogen, op basis van spiegels en farmacokinetiek. Een andere strategie is het switchen naar een extended half-life factor VIII-concentraat omdat deze ook bij remmerpatiënten een langere halfwaardetijd hebben. (consensus) |
|
C |
Patiënten met zeer hoge remmer titers (>200 BU) zijn het meest gebaat zijn bij een intensiever schema van hoge dosis ITI. (consensus) |
|
C |
Onderbreking van ITI moet vermeden worden omdat het de kans op het bereiken van tolerantie vermindert. (Lenk, 2000) |
|
C |
Na het starten van ITI wordt meestal in de eerste 2-4 weken een stijging van de remmertiter gezien (anamnestische respons genoemd).Dit is het moment om de piek titer vast te stellen. (consensus) |
|
C |
ITI moet worden gecontinueerd zo lang er een dalende trend is te zien in de remmer titer (in ieder geval 20% daling in 6 maanden). Als deze criteria niet gehaald worden moet intensivering van het ITI schema overwogen worden. (consensus) |
|
C |
De maximale duur van een ITI behandeling wordt over het algemeen gezien als 33 maanden omdat na deze periode de kans op succes extreem klein wordt geacht. (consensus) |
|
C |
Een remmer wordt pas echt als verdwenen (negatief) beschouwd bij aanhoudend negatieve remmer testen, een opbrengst van > 66% van verwacht, en een halfwaardetijd van minimaal 7 uur voor standaard half-life factor VIII en minimaal 14 uur voor standaard half-life factor IX. Voor extended half-life concentraten wordt de kortst gepubliceerde halfwaardetijd als een minimale halfwaardetijd aangehouden. (consensus) |
|
C |
Na het bereiken van tolerantie met hoge dosis ITI moet de behandeling langzaam afgebouwd worden tot een normale profylactische dosis. (Collins, 2017; Carcao, 2019) |
|
B |
Bij patiënten met ernstige hemofilie A met een remmer die geen of onvoldoende effect hebben op ITI behandeling zijn positieve resultaten beschreven van behandeling met immunomodulerende therapie. Combinatie van immunomodulatie met ITI lijkt het meest effectief te zijn. (Giulino, 2007; Franchini, 2008; Collins, 2009) |
|
B |
Bij patiënten met matig ernstige of milde hemofilie A met een remmer is beschreven dat bij 70% de remmer spontaan verdwijnt als er geen blootstelling is aan factor VIII-concentraat. Echter, bij 40% van deze patiënten komt de remmer terug bij gebruik van factor VIII-concentraat (anamnestische reactie) en blijkt er dus geen sprake te zijn van tolerantie. (van Velzen, 2015) |
|
B |
Bij patiënten met matig ernstige of milde hemofilie A met een remmer is beschreven dat de remmer kan verdwijnen na monotherapie met rituximab, ook zonder voorafgaande behandeling met ITI. (van Velzen, 2015; Kempton, 2012; Dunkley, 2006) Er blijkt echter niet bij iedereen sprake te zijn van tolerantie, aangezien na rechallenge met factor VIII-concentraat een anamnestische reactie kan optreden. |
|
B |
Bij patiënten met hemofilie B en een remmer is ITI veelal niet succesvol en treedt vaak anafylaxie op al dan niet met ook nefrotisch syndroom. Immunomodulerende therapie, al dan niet na of in combinatie met ITI was in een beperkt aantal patiënten succesvol. (Santoro, 2018; Collins, 2013; Rocino, 2014; Young, 2019) |
Samenvatting literatuur
Resultaten
Immuuntolerantie inductie (ITI)
Omdat een remmer interfereert met de normale behandeling van ernstige hemofilie hebben remmer patiënten een hogere mortaliteit (Walsh, 2015) en morbiditeit (Morfini, 2007) en zelfs als kind al meer gewrichtsschade dan patiënten zonder remmers (Monahan, 2011). Doordat primaire profylaxe niet meer effectief is en ze daardoor een groot risico lopen op irreversibele gewrichtsschade, zijn alle patiënten met ernstige A en een remmer in principe kandidaat voor ITI (immuun tolerantie inductie) therapie. Deze behandeling is erop gericht om de immuunrespons tegen factor VIII ‘uit te laten doven’ door middel van aanhoudende regelmatige toediening van factor VIII.
Predictoren van succes van ITI: Wanneer starten?
Sinds de introductie in de jaren 70 is het succes percentage ongeveer gelijk gebleven, rond 60-70%. Ter illustratie: in het verslag van het internationale ITI register van de ISTH werden 158 patiënten gevolgd waarbij in 67,7% tolerantie werd bereikt en in 7,6% een partiële respons (Mariani, 1994). In de gerandomiseerde internationale ITI (n=115) studie werd 69,7% tolerant en had 4,5% een partiële respons (Hay, 2012).
Uit data van de North American Immune Tolerance Registry (NAITR) leek het of de remmer titer pre-ITI een zeer belangrijke predictor van ITI succes was: in 164 remmerpatiënten werd van patiënten met een titer van max. 10 BU bij de start van ITI 83% tolerant, vs. 40% van de patiënten met een hogere titer bij de start van ITI (DiMichele, 2002). Een kleine studie van Nakar et al. suggereerde, dat direct beginnen met ITI geassocieerd was met het bereiken van tolerantie: tolerantie werd bereikt in 22/23 (96%) van hoge titer remmerpatiënten, die binnen een maand na diagnose waren begonnen met ITI vs. 7/11 (64%) van de patiënten die later waren begonnen (Nakar, 2015). Dit is ook een reden om snel te starten met ITI (Carcao, 2019). Daarom wordt tegenwoordig door veel behandelaren direct na het bevestigen van de diagnose begonnen met ITI, onder andere in de PedNet centra waar ITI werd gestart mediaan 1,3 maanden na diagnose (interquartile range (0,2-8,7 maanden) (Mancuso, 2017) en in de UK (Collins, 2017). Er is gepostuleerd dat met een directe start van ITI misschien de maturatie van de immuunrespons met vorming van langlevende plasmacellen wordt voorkómen (Liu, 2016).
Productkeuze voor ITI
Meestal wordt voor ITI het product gebruikt waarop de remmer is ontwikkeld (Mannucci, 2012), maar er zijn verschillende rapporten, die suggereren dat VWF bevattend uit plasma vervaardigd factor VIII succesvol gebruikt kan worden voor ITI bij patiënten die een remmer hebben ontwikkeld op recombinant factor VIII. Sommigen claimen een hoger succes percentage (Kreuz, 2016; Oldenburg, 2014; Santagostino, 2019; Rangarajan, 2014). Omdat een gerandomiseerde studie ontbreekt en een pooled meta analyse geen positief effect van VWF bevattende factor VIII-concentraten liet zien (van Velzen 2014), kunnen hierover nog geen aanbevelingen gedaan worden. Recent werd in een retrospectieve analyse gesuggereerd dat factor VIII-Fc (Elocta®) een goed resultaat zou hebben bij (rescue) ITI, dit moet nog bevestigd worden in andere studies (Carcao, 2018).
ITI regime/behandeling
Van oudsher worden er twee regimes onderscheiden:
De internationale ITI studie, een multicenter RCT in 115 kinderen met ernstige hemofilie A, titer 5-200 BU, vergeleek het effect van lage dosis ITI (3x per week 50 E/kg) met hoge dosis ITI (200 E/kg/dag) (Hay, 2012). Het succespercentage was 74,2% (69,7% volledig tolerant, 4,5% partiële respons), in beide armen. Wel had de hoge dosis arm een snellere respons (tijd tot negatieve titer mediaan 4,5 vs. 9,2 maanden) en significant minder bloedingen ten tijde van een positieve remmer titer (0,28 bloedingen/maand vs. 0,62 bloedingen per maand; p < 0.001) (Hay, 2012). Voor patiënten met een remmer titer van > 100 BU geldt dit in mindere mate en moet rekening worden gehouden met het optreden van (subklinische) bloedingen.
Gebaseerd op het klinische beloop en expert opinion (Hay, 2006; Valentino, 2015; Ter Avest, 2010; Carcao, 2019) wordt geadviseerd om patiënten met lage titer remmers (titer steeds < 5 BU) geen ITI te geven maar gewone profylaxe te geven. In Nederland komt dit neer op ± 25 E/kg 3x per week of om de dag. In het PedNet cohort van 260 remmerpatiënten is gezien, dat ongeveer 50% van de lage titer remmers zich ontwikkelt tot een hoge titer remmer (≥ 5 BU) na het starten van ITI. Dit was afhankelijk van het gendefect (null mutaties OR 2,6 (CI 1,0-6,5), een positieve familieanamnese voor remmers (OR 7,2 (CI 1,8-28,4) en de hoge dosis ITI. Voor patiënten die ≥ 100E/kg/dag kregen, was de OR 3,9 (CI 1,5-10,0) (Mancuso, 2017). Alleen bij progressie van de remmer titer tot > 200 BU moet hoge dosis ITI overwogen worden.
Bij patiënten met remmers met een piek titer tussen de 5 en 200 BU wordt geadviseerd lage dosis ITI toe te passen. Dit geldt zeker voor de groep met de beste prognose (piek titer tot max. 40 BU). In een Nederlands cohort van 21 patiënten werden alle 17 patiënten met een piek titer van max. 40 BU tolerant met lage dosis ITI (Ter Avest, 2010). Deze patiënten hadden ook in de internationale ITI studie een zeer goede prognose: daar werd een twee keer zo hoge respons op ITI gezien bij patiënten met een max. piek titer van < 36 BU (50% tolerant, vs. 25% in patiënten met een hogere pre-ITI titer (Hay, 2012). Ook patiënten met een remmer titer tussen de 40 en 200 BU hebben een goede kans om tolerant te worden. In deze groep wordt derhalve geadviseerd te starten met lage dosis ITI.
Bij patiënten met een piek titer tot 200 BU die bloedingen vertonen, kan gekozen worden uit een aantal strategieën om bloedingen te voorkomen:
Hoewel er geen data zijn over patiënten met zeer hoge remmer titers voor ITI (> 200 BU) is de algemene consensus, dat deze patiënten het meest gebaat zijn bij een intensiever schema van hoge dosis ITI. Met de beschikbaarheid van emicizumab ter preventie van bloedingen bij patiënten met een remmer wordt het argument om een hoge dosis ITI te geven ter preventie van bloedingen relatiever. Een andere strategie zou kunnen zijn om primair emicizumab profylaxe te starten, eventueel in combinatie met laagfrequente toediening van factor VIII met het doel uiteindelijk immuuntolerantie te bewerkstelligen (Carcao, 2019). Onderbreking van ITI moet vermeden worden, omdat het de kans op het bereiken van tolerantie vermindert (Lenk, 2000).
Monitoring van ITI
In de UKHCDO richtlijn wordt geadviseerd na start met ITI de remmer titer gedurende 1 maand wekelijks te meten om de piek titer vast te stellen en daarna maandelijks (UKHCDO guideline, 2017; www.ukhcdo.org). De NVHB adviseert om in de 2-4 weken na het starten van ITI de remmer titer te controleren om de piek titer vast te stellen. Verschillende richtlijnen suggereren om de ITI te intensiveren als de anamnestische respons heftig is. In patiënten die met lage dosis ITI behandeld worden als een piek > 40 BU gezien wordt, en bij een titer >200 BU als er hoge dosis ITI gegeven wordt (Collins, 2013, 2017; Valentino, 2015) .
ITI moet worden gecontinueerd zo lang er een dalende trend is te zien in de remmer titer (in ieder geval 20% daling in 6 maanden). NB infecties kunnen een (tijdelijke) stijging van de remmer titer tot gevolg hebben. In dit geval is het raadzaam de remmer titer ± 2 weken na resolutie van de infectie te herhalen. Als patiënten op een hoge dosis ITI onvoldoende respons laten zien, moet aanpassing van het ITI regime en/of immunomodulatie overwogen worden.
De maximale duur van een ITI behandeling wordt over het algemeen gezien als 33 maanden, omdat na deze periode de kans op succes extreem klein wordt geacht.
Zodra de remmer titer rond de 1 BU is, moet overwogen worden om opbrengsten na 15-20 min en na 24 uur te meten. Dit geeft een indruk van de bescherming tegen bloedingen en geeft ook de mogelijkheid om de halfwaardetijd te schatten, bijvoorbeeld met behulp van programma’s als WAPPS.org of OPTI-CLOT .
Afbouwen ITI bij negatieve remmer titer
Een remmer wordt pas echt als verdwenen (negatief) beschouwd bij aanhoudend negatieve remmertesten, een opbrengst van > 66% van verwacht en een halfwaardetijd van minimaal 7 uur voor standaard half-life factor VIII en minimaal 14 uur voor standaard half-life factor IX. Voor extended half-life concentraten wordt de kortst gepubliceerde halfwaardetijd als een minimale halfwaardetijd aangehouden. Voor lage dosis ITI is het waarschijnlijk niet nodig om de dosis aan te passen (te verlagen), omdat de halfwaardetijd eerst toch verkort is en de patiënt langzaam uit zijn dosis zal groeien.
Bij hoge dosis ITI moet de behandeling langzaam afgebouwd worden om een relapse van de remmer maar ook bloedingen te voorkomen. Bij het bereiken van 24 uurs-spiegels van minimaal 1% gedurende 2 opeenvolgende maanden, kan overwogen worden om de dosis aan te passen (stap 1, met maximale stappen van 50% o.b.v. ampulgrootte). Als de spiegels goed blijven, kan per ± 3 maanden een dosisreductie doorgevoerd worden. Als de dosis ± 50 E/kg/dag is, kan de frequentie van ITI verlaagd worden naar om de dag (Collins, 2017; Carcao, 2019).
Gebruik van immunomodulerende therapie met name rituximab ter eliminatie van remmers
Rituximab is een chimeer muizen-humaan monoclonaal IgG1 antilichaam tegen CD20 antigeen op het oppervlak van B lymfocyten. Binding van rituximab aan B lymfocyten resulteert in celdood. Dit leidt tot een snelle en langdurig aanhoudende eliminatie van B lymfocyten. De gebruikelijke dosering is 375 mg/m2 per week gedurende 4 weken.
Rituximab wordt in het algemeen gebruikt als tweedelijns behandeling tegen inhibitors bij mensen die resistent zijn voor behandeling met ITI. Er zijn geen gerandomiseerde, gecontroleerde trials die de effectiviteit en veiligheid van rituximab hebben onderzocht. Er zijn uitsluitend case reports en case series gepubliceerd (Jiangl, 2017). Behalve rituximab worden ook andere medicamenten, zoals corticosteroiden, met name dexamethason, intraveneus gammaglobuline (IVIG), ciclosporine en mycofenolaat mofetil (MMF) gebruikt voor immunomodulatie om het effect van ITI te vergroten dan wel om zonder combinatie met ITI de remmer te doen verdwijnen (Beutel, 2009).
Er zijn 3 systematische reviews gepubliceerd, namelijk door Giulino in 2007, Franchini in 2008 en Collins in 2009. Francini includeerde 29 studies met 49 patiënten met erfelijke hemofilie A en B met inhibitors die resistent waren tegen ITI. De gemiddelde leeftijd was 14 jaar (range 1-70 jaar). Dertig waren kind of adolescent. De mediaan van de inhibitor titer was 30 BU (range 2-1562 BU). Drieëntwintig patiënten hadden ernstige hemofilie A, 5 ernstige hemofilie B. Van de patiënten had 66,7% eerdere ITI gehad. De dosering rituximab was 375 mg/m2 per week. Het mediane aantal giften rituximab was 4,7 (range 4 tot 11). Vierentwintig van de 49 patiënten kregen tevens hoge doseringen immuunglobulinen, immunosuppressiva of plasmaferese/immunoabsorbtie. Er werd een aanhoudende remissie bereikt bij 26 van de 49 patiënten (53,1%) bij een follow-up van 3 tot 34 maanden. De mediane tijd tot respons was 3 maanden (range 1-15 maanden). Er waren 7 relapsen, 2 bereikten weer een complete remissie na herhaalde behandeling met rituximab. Eén patiënt kreeg een onderhoudsbehandeling rituximab. Patiënten met matige en milde hemofilie hadden een betere respons dan die met ernstige hemofilie. Gelijktijdige behandeling met factor VIII-concentraat en immunosuppressiva had een positief effect op de behandeling. Het effect van immunosuppressiva was alleen aanwezig bij gelijktijdige behandeling met factor VIII-concentraat. Drie van de 5 ernstige hemofilie B patiënten hadden een complete remissie. Eén milde hemofilie B patiënt had geen respons (Franchini, 2008).
Giulino beschreef 8 kinderen met erfelijke hemofilie met een remmer die met rituximab zijn behandeld. Vijf kinderen (63% hadden een initiële respons, maar 2 kregen een relapse na 11 en 8 maanden. Eén van hen respondeerde na een tweede behandeling met rituximab (Giulino, 2007).
Collins beschrijft een cohort van 15 hemofilie A patiënten met een remmer die met rituximab zijn behandeld. Alle patiënten waren tevoren zonder succes behandeld met ITI. De mediane leeftijd was 6 (range 2-37) jaar. De piek remmer titer was 331 (range 28-6000) BU. Zeven patiënten waren van Afrikaanse origine, 7 waren Kaukasisch en 1 was afkomstig uit India. De mediane follow-up was 108 weken (range 26-292 weken). De dosering rituximab was 375 mg/m2, standaard werden 4 kuren gegeven. Twaalf van de 15 patiënten kregen ook factor VIII-concentraat. Vier patiënten kregen tevens immunosuppressieve behandeling. Zes patiënten bereikten een negatieve titer, waarvan 3 een relapse kregen. Vier patiënten hadden een partiële respons met klinische verbetering en 5 patiënten hadden geen respons. In totaal hadden slechts 2/15 patiënten een langdurige complete respons. Een significante en langdurig aanhoudende klinische verbetering werd bij 7/15 patiënten gezien. Alle 3 patiënten die geen factor VIII als comedicatie kregen, bereikten geen complete respons. Van de 3 patiënten met een relapse kregen 2 een herhaalde behandeling met rituximab en factor VIII-concentraat en zij bereikten weer een complete remissie. De 3e patiënt kreeg alleen rituximab en bereikte geen remissie (Collins, 2009).
Er werden verder diverse case histories en kleine series beschreven bij patiënten met ernstige en niet ernstige hemofilie A met soms goed resultaat en soms ook minder goed resultaat van behandeling met rituximab al dan niet in combinatie met ITI en/of andere immunosuppressieve behandeling (Chuansumrit, 2007; Curry, 2006; Ranta, 2011; Leissinger, 2014).
Gezien de bevindingen in de literatuur is het algemene advies om bij patiënten met ernstige hemofilie A en een remmer die niet of onvoldoende reageert op behandeling met ITI immunomodulerende therapie, met name rituximab al dan niet in combinatie met andere immunomodulerende medicamenten, voor te schrijven. Bij ernstige hemofilie A lijkt een combinatie van rituximab met ITI effectiever te zijn dan monotherapie met rituximab (Laros-van Gorkom, 2014). Bij niet ernstige hemofilie A is monotherapie met rituximab wel effectief, ook zonder dat eerst ITI wordt gegeven.
Niet ernstige hemofilie A
Bij patiënten met matig ernstige en milde hemofilie en een remmer is de morbiditeit en mortaliteit ook verhoogd (Eckhardt, 2015).
Het retrospectieve Europese INSIGHT cohort dat 101 remmerpatiënten met niet ernstige hemofilie includeerde liet zien dat in de meerderheid van de patiënten (71 %; 72/101) de remmer niet meer aantoonbaar was, hetzij spontaan (70 %, 51/73), hetzij na behandeling, in de vorm van ITI of immunosuppressie (75 %, 21/28) (van Velzen, 2015). Werkelijke tolerantie (geen anamnestische reactie na een rechallenge met factor VIII-concentraat, nadat de remmer verdwenen was) was aantoonbaar in 64 % (30/47) van de patiënten die een rechallenge hebben ondergaan. Bij patiënten met een hoge titer remmer was werkelijke tolerantie geassocieerd met eradicatie behandeling (RR 2,3, 95 % CI 1,3–4,3), vergelijken met de groep die geen eradicatie behandeling kreeg.
Er zijn 2 case series (Kempton, 2012; Dunkley, 2006) en 1 retrospectieve cohort studie (Lim, 2014) gepubliceerd waarin specifiek rituximab behandeling van remmerpatiënten met niet ernstige (dus matig ernstige of milde) hemofilie A worden beschreven. Dunkley beschrijft 3 patiënten met milde hemofilie A waarbij de remmer verdwijnt met rituximab. Bij de eerste patiënt waren vanwege het tweemaals terugkeren van de remmer 3 kuren met rituximab nodig en is de remmer onder controle met elke 3 maanden een rituximab dosis. Van de tweede patiënt is onbekend of hij tolerant is voor factor VIII omdat er nog geen rechallenge heeft plaatsgevonden. De derde patiënt is tolerant met een follow-up van 6 maanden. Kempton beschrijft 36 patiënten met niet ernstige hemofilie A waarvan er 6 met rituximab worden behandeld, bij allen is er een positief effect, waarbij de remmer verdwijnt, maar er is geen informatie beschikbaar over tolerantie na een rechallenge met factor VIIII-concentraat. Lim beschrijft 9 patiënten, ten dele overlappend met Kempton, met niet ernstige hemofilie A die met rituximab worden behandeld en waarbij de remmer bij alle patiënten verdween. Er werd in dit cohort geen behandeling met ITI, corticosteroiden of andere immunosuppressiva gegeven. De mediane responstijd was 95 dagen (12-278 dagen), de mediane follow-up tijd was 56 maanden (range 13-139 maanden). Acht patiënten werden opnieuw met factor VIII behandeld, waarvan er 2 een recidief kregen in een post-operatieve setting.
Hemofilie B
Met betrekking tot patiënten met hemofilie B met een remmer is recent een review artikel gepubliceerd door Santoro et al. (Santoro, 2018). Er zijn uitsluitend case histories en kleine series patiënten beschreven. Er is geen prospectief onderzoek gepubliceerd. Bij hemofilie B gaat het optreden van remmers frequent gepaard met overgevoeligheid of anafylaxie tegen toegediend factor IX waarbij vaak een nefrotisch syndroom optreedt. ITI is veelal niet succesvol. Er is een aantal patiënten beschreven, waarbij behandeling met rituximab, soms ook gecombineerd met corticosteroiden, mycofenolaat mofetil of IVIG succesvol was in de eradicatie van de remmer en het herstel van tolerantie voor factor IX-concentraat. Het is daarom te overwegen om bij patiënten met hemofilie B met een remmer ITI upfront of na falen van ITI alleen te combineren met immunomodulerende behandeling (rituximab, corticosteroiden, mycofenolaat mofetil of IVIG) (Beutel 2009; Santoro 2018). Dit beleid wordt ook door anderen aangeraden (Collins, 2013; Rocino, 2014; Young, 2019).
Referenties
Bewijskracht literatuur
De bewijskracht uit de literatuur is laag. Er is slechts één direct vergelijkende, gerandomiseerde studie gedaan naar het bereiken van tolerantie bij patiënten met ernstige hemofilie A en een remmer waarbij 2 doseringsschema’s werden vergeleken (Hay, 2012). Verder is er alleen sprake van cohortonderzoeken, case histories en consensus op basis van expert opinion.
Level 2, consistent.
Zoeken en selecteren
Er werd voor de behandeling met ITI geen systematische search gedaan. Er werd hierbij gebruik gemaakt van bekende richtlijnen en de daarin vermelde relevante referenties en bij de auteurs bekende relevante publicaties.
Voor de behandeling met immuunmodulerende medicamenten werd een literatuursearch gedaan met als zoektermen hemophilia, inhibitor, rituximab and immunomodulation. Verder werden bij de auteurs bekende relevante publicaties geraadpleegd.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Er zijn geen evidence tabellen.
Overwegingen
Er werd in de internationale ITI studie (Hay, 2012) geen verschil gevonden in het percentage bereiken van immuuntolerantie tussen patiënten die met het lage doseringsschema waren behandeld en patiënten die met het hoge doseringsschema waren behandeld. Wel werden minder bloedingen gezien bij het hoge doseringsschema. Gezien de kosten van de ITI behandeling en de belasting voor de patiënt is er voor gekozen om bij alle patiënten met een remmertiter tot 200 BU te starten met lage dosis ITI.
Voor het moment van starten van ITI, de keuze van het gebruikte factorconcentraat bij ITI, het te gebruiken doseringsschema bij ITI afhankelijk van de remmertiter en de monitoring van de remmertiter bij ITI is geen eensluidende conclusie uit de beschikbare literatuur te trekken. De conclusies en adviezen zijn gebaseerd op consensus in de diverse richtlijnen.
Met de komst van emicizumab is de preventie en behandeling van bloedingen bij patiënten met hemofilie A en een remmer enorm veranderd. Hierdoor komt ook de noodzaak tot het doen verdwijnen van een remmer in een ander licht te staan. In de komende tijd zal onderzocht moeten gaan worden wat de voor- en nadelen zijn van het bereiken van tolerantie door ITI en immuunmodulerende therapie bij patiënten met hemofilie A en een remmer.
Hoe is de preventie en behandeling van bloedingen bij een patiënt met een remmer?
Aanbevelingen
Onderbouwing
Inleiding
Preventie van bloedingen en daarmee het voorkomen van gewrichtsschade is het primaire doel van de behandeling van hemofilie. Sinds het beschikbaar komen van emicizumab voor de preventie van bloedingen bij patiënten met hemofilie A met een remmer is dit medicament de voorkeurs-behandeling geworden bij deze patiëntcategorie (Oldenburg, 2017). Hiermee zijn andere behandelingen ter preventie van bloedingen bij patiënten met hemofilie A en een remmer naar de achtergrond verdrongen. Zij kunnen eventueel nog wel relevant blijken bij patiënten die resistent worden tegen emicizumab. Het beleid met betrekking tot het starten en voortzetten van ITI zal mogelijk worden aangepast op basis van nieuw ter beschikking komende onderzoeksresultaten.
Conclusies
|
SORT grade |
Conclusie |
|
A |
Emicizumab is effectiever dan bypassing agents in de preventie van bloedingen bij patiënten met hemofilie A en een remmer. (Oldenburg, 2017; Young, 2019) |
|
B |
Profylactische behandeling met bypassing agents vermindert het aantal bloedingen bij patiënten met hemofilie A en een remmer. (Oldenburg, 2017; Young, 2019) |
|
C |
Door de langere halfwaardetijd van FEIBA® heeft dit middel de voorkeur boven geactiveerd recombinant factor VII bij de preventie van bloedingen bij patiënten met hemofilie A en een remmer. (consensus) |
|
C |
Intensiveren van het ITI regime vermindert de kans op bloedingen bij patiënten met hemofilie A en een remmer. (Hay, 2012) |
|
C |
ITI met extended half-life factor VIII-concentraten vermindert mogelijk de kans op bloedingen bij patiënten met hemofilie A en een remmer. (Carcao, 2018) |
|
C |
Patiënten met hemofilie B en een remmer met bloedingen kunnen ter preventie van bloedingen profylactisch met geactiveerd recombinant factor VII worden behandeld. (Kavakli, 2017; Scharrer, 1999) |
|
A |
Bloedingen bij patiënten met hemofilie A en een remmer onder emicizumab kunnen met geactiveerd recombinant factor VII worden behandeld. (Oldenburg, 2017; Young, 2019) |
|
A |
Bloedingen bij patiënten met hemofilie A en een remmer die niet met emicizumab worden behandeld kunnen met FEIBA® of geactiveerd recombinant factor VII worden behandeld. (Astermark, 2007; Santagostino, 2009; Collins, 2013; Ljung, 2019) |
|
C |
Bij patiënten met hemofilie A en een remmer wordt een synergistisch effect gezien van de toediening van een bypassing agent (FEIBA® of geactiveerd recombinant factor VII) en een hoge dosis FVIII (50 E/kg). (Livnat, 2017) |
Samenvatting literatuur
Resultaten
Preventie van bloedingen bij patiënten met hemofilie A en een remmer
Emicizumab
Emicizumab (Hemlibra®) is een bispecifiek gehumaniseerd monoclonaal antilichaam dat een brug vormt tussen geactiveerd factor IXa en factor X, waarmee het de functie overneemt van ontbrekend factor VIII bij mensen met hemofilie A. Omdat emicizumab een geheel ander molecuul is dan factor VIII is het ongevoelig voor remmers tegen factor VIII.
Emicizumab is gebleken effectief te zijn in de preventie van bloedingen bij patiënten met hemofilie A met een remmer (HAVEN 1 studie, volwassenen (Oldenburg, 2017); HAVEN 2 studie kinderen, (Young, 2019)). In de HAVEN 1 studie werden 109 patiënten met hemofilie A en een remmer van 12 jaar of ouder onderzocht. In 35 patiënten die profylactisch behandeld werden met emicizumab (groep A), was het aantal bloedingen per jaar (annualized bleeding rate, ABR) 2,9 (95% CI 1,7-5,0) in vergelijking met 23,3 (95% CI 12,3-43,9) in 18 patiënten die geen profylaxe kregen (groep B). Dit was een vermindering van 87% (p < 0.001) ten gunste van emicizumab. 22 patiënten in groep A (63%) hadden geen enkele bloeding in vergelijking met 1 patiënt (6%) in groep B. In groep C (24 patiënten) resulteerde profylaxe met emicizumab in een 79% lagere bloedingsfequentie dan tevoren onder profylaxe met bypassing agents (p < 0.001). De meest frequente bijwerking van emicizumab was een reactie op de injectie plaats. Trombotische microangiopathie en trombose werden gezien in 2 patiënten, die naast de emicizumab behandeld waren met meerdere infusies van FEIBA® bij doorbraakbloedingen (Oldenburg, 2017).
In de HAVEN 2 studie werden 85 patiënten met hemofilie A en een remmer jonger dan 12 jaar onderzocht, die tevoren on demand of profylactisch waren behandeld met bypassing agents. Er werden 3 doseringen emicizumab vergeleken (groep A 1,5 mg/kg/week; groep B 3 mg/kg/2 weken; groep C 6mg/kg/4 weken). In groep A was de ABR 0,3 (95CI 0,17-0,5) en 77% had in het geheel geen bloedingen. In 15 patiënten werd er vergeleken met een voorgaande periode van profylaxe met bypassing agents. Emicizumab verminderde de ABR met 99% (95% CI 97,4-99,4). In groep B (n=10) was de ABR 0,2 en in groep C (n=10) was de ABR 2,2. De meest voorkomende bijwerkingen waren nasofaryngitis en reacties op de injectieplaats. Er werd geen trombose gezien. Twee patiënten ontwikkelden antistoffen tegen emicizumab, in 1 patiënt resulteerde dit in verlies van de effectiviteit en in de andere verdwenen de antistoffen in de loop van de tijd (Young, 2019).
Uit de HAVEN 1 en 2 studies kan geconcludeerd worden dat profylaxe met emicizumab zeer effectief is in de preventie van bloedingen bij patiënten met hemofilie A en een remmer. Er is geen prospectief onderzoek gedaan met een vergelijking met bypassing agents. Wel werd in beide studies een intrapatient vergelijking gemaakt met een voorgaande periode van profylaxe met bypassing agents, waarbij een sterke reductie van de ABR werd gezien.
Tijdens de behandeling met emicizumab kan bij bloedingen factor VIII-concentraat worden bijgegeven. Dit zal in aanwezigheid van een remmer echter niet effectief zijn. Daarom zal bij een bloeding bij een patiënt met hemofilie A en een remmer een bypassing agent gegeven moeten worden. Bij de combinatie van emicizumab met hoge doseringen geactiveerd protrombinecomplexconcentraat (APCC, FEIBA®) zijn ernstige bijwerkingen gezien, met name trombotische microangiopathie.
De combinatie van emicizumab en FEIBA® moet daarom zo mogelijk vermeden worden. In geval van ernstige refractaire bloedingen, die niet onder controle komen met geactiveerd recombinant factor VII, kan overwogen worden FEIBA® te geven in een lagere dosering, gezien het risico van trombotische microangiopathie bij doseringen van >,100E/kg en gedurende meer dan 24 uur.
Bypassing agents (FEIBA® en geactiveerd recombinant factor VII)
Er is slechts beperkte literatuur over gebruik van bypassing agents ter preventie van bloedingen bij patiënten met hemofilie en remmers. Voor zowel gebruik van FEIBA® (Leissinger, 2007; Antunes, 2014; Leissinger, 2011) als voor geactiveerd recombinant factor VII (Konkle, 2007) werd in retrospectief en prospectief onderzoek de effectiviteit en veiligheid aangetoond in de reductie van het aantal bloedingen bij patiënten met remmers. Er is geen direct vergelijkend onderzoek gedaan tussen beide ’bypassing agents’ in de profylaxe van bloedingen. Bij de behandeling van bloedingen in patiënten met remmers werd geen verschil in effectiviteit gevonden (Astermark, 2007). Op basis van deze gegevens wordt profylaxe met een bypassing agent algemeen geadviseerd in diverse richtlijnen (Santagostino, 2009; Collins, 2013; Ljung, 2019) met een wisselende voorkeur voor een van beide middelen.
Intensiveren van ITI behandelschema
In de internationale ITI studie, waarin een vergelijking werd gemaakt tussen een lage en een hoge dosis ITI, bleek dat er bij de hoge dosering minder bloedingen optraden (Hay, 2012). Op grond hiervan kan overwogen worden het ITI behandelschema te intensiveren met als eerste stap het verhogen van de frequentie van de infusies om het optreden van bloedingen te verminderen.
Preventie van bloedingen bij patiënten met hemofilie B en een remmer
Omdat het werkingsmechanisme van geactiveerd recombinant factor VII onafhankelijk is van factor VIII of IX, kunnen ook patiënten met hemofilie B en een remmer met bloedingen en ter preventie van bloedingen met geactiveerd recombinant factor VII worden behandeld (Kavakli, 2017; Scharrer, 1999).
Behandeling van bloedingen bij patiënten met hemofilie A en een remmer
Over het algemeen kunnen bloedingen bij deze patiënten behandeld worden met bypassing agents (recombinant geactiveerd factor VII of FEIBA®). Bij lage titer remmers kan ook nog behandeld worden met een hoge dosis factor VIII (140 E/kg), waarbij de opbrengst gecontroleerd moet worden en rekening moet worden gehouden met een sterk verkorte halfwaardetijd. In de praktijk is het resultaat van een hoge dosis factor VIII verwaarloosbaar bij patiënten met een remmer boven de 3 BU. De oorspronkelijke formule gehanteerd om de remmer te neutraliseren (i.e. 2x lichaamsgewicht in kg x 80 x (100-hematocriet in %) x remmertiter in BU/ml :100) geeft niet altijd de juiste resultaten.
Bij patiënten met hoge titer remmers is het soms moeilijk om een bloeding (snel) tot staan te brengen. In vitro, en beperkt in vivo, zijn er aanwijzingen dat factor VIII en bypassing agents een synergistisch effect hebben. Dit kan bij patiënten die naast ITI profylaxe met bypassing agents krijgen uitgetest worden met behulp van trombinegeneratiebepaling. De groep van professor Kenet in Israël liet in een single center studie zien dat in het bloed van 15 patiënten (leeftijd 1-72) met hoge titer remmers (range 4-620) de trombinegeneratie significant toenam als bypassing agents met factor VIII gecombineerd werden, waarbij de combinatie FEIBA® en factor VIII een hogere trombinegeneratie gaf dan de combinatie recombinant geactiveerd factor VII en factor VIII. Vervolgens werden 64 bloedingen in 7 patiënten behandeld met de combinatie FEIBA® (50-75 E/kg) en FVIII (50-100 E/kg). De bloeding kwam in 85% tot staan met 1 infusie. Bij in totaal 332 gecombineerde infusies van bypassing agent en factor VIII werden geen bijwerkingen gezien (Livnat, 2017).
Bij patiënten met hoge titer remmers en refractaire dan wel levensbedreigende bloedingen kan men overwegen om bypassing agents alternerend te geven. De groep uit Israël deed in 2009 eerst een in vitro studie naar de effecten van gecombineerde toediening van FEIBA® en recombinant geactiveerd factor VII op trombinegeneratie in vijf patiënten met een hoge titer remmer (range 8-1300 BU). Vervolgens behandelden zij 400 bloedingen met een alternerend regime van 30-70 microgr/kg recombinant geactiveerd factor VII (Novoseven®) en 20-30E/kg FEIBA®. Hierbij werd de halfwaardetijd in acht genomen: de recombinant geactiveerd factor VII werd zes uur na de FEIBA® gegeven en de FEIBA® twee uur na de recombinant geactiveerd factor VII. Er werden geen bijwerkingen, met name geen tromboses gezien (Martinowitz, 2009). Buiten de groep in Israël werden er geen publicaties over deze strategie gevonden.
Behandeling van bloedingen bij patiënten met hemofilie B en een remmer
Het is duidelijk dat ook patiënten met hemofilie B en remmers behandeld kunnen worden met recombinant geactiveerd factor VII in dezelfde dosis als bij hemofilie A. Het gebruik van FEIBA® wordt ontraden in verband met het risico op een allergische reactie op de factor IX die in FEIBA® zit (Collins, 2013).
Referenties
Bewijskracht literatuur
Er zijn 2 prospectieve onderzoeken gedaan naar de effectiviteit van emicizumab ter preventie van bloedingen bij patiënten met ernstige hemofilie A met remmers (Oldenburg, 2017; Young, 2019). In beide onderzoeken werd niet gerandomiseerd vergeleken met standaardtherapie. Hierdoor is de bewijskracht beperkt.
Level 2, consistent.
Voor de behandeling van bloedingen bij patiënten met hemofilie A met een remmer werd een gerandomiseerd vergelijkend onderzoek gedaan tussen FEIBA® en geactiveerd recombinant factor VII (Astermark, 2007). Er werd hierbij geen verschil in effectiviteit tussen beide middelen gevonden.
Level 1, consistent.
Verder zijn er geen direct vergelijkende prospectieve gerandomiseerde onderzoeken gedaan zodat de bewijskracht beperkt blijft.
Zoeken en selecteren
Er werd geen systematische search gedaan. Er werd gebruik gemaakt van bekende richtlijnen en de daarin vermelde relevante referenties en bij de auteurs bekende relevante publicaties.
Zoekverantwoording
Niet van toepassing.
Evidence tabellen
Er zijn geen evidence tabellen.
Overwegingen
Voor de behandeling van bloedingen bij patiënten met hemofilie A met een remmer werd een gerandomiseerd onderzoek gedaan naar de effectiviteit van FEIBA® en geactiveerd recombinant factor VII. Hierbij werd geen verschil in effectiviteit gevonden. Voor de profylaxe van bloedingen bij deze patiënten werd geen vergelijkend onderzoek gedaan. Aangezien FEIBA® een langere halfwaardetijd heeft dan geactiveerd recombinant factor VII, ±6 uur respectievelijk 2 uur, gaat bij gebruik van een bypassing agent als profylaxe van bloedingen de voorkeur uit naar FEIBA®. Door de langere halfwaardetijd is profylaxe met FEIBA® waarschijnlijk effectiever dan met geactiveerd recombinant factor VII.
In de internationale ITI studie waarin een vergelijking werd gemaakt tussen een lage en een hoge dosis ITI bleek dat er bij de hoge dosering minder bloedingen optraden (Hay, 2012). Op grond hiervan kan overwogen worden het ITI behandelschema te intensiveren met als eerste stap het verhogen van de frequentie van de infusies om het optreden van bloedingen te verminderen.
De pathofysiologische verklaring van de trombotische complicaties gezien bij patiënten waarbij emicizumab gecombineerd werd met hoge dosis FEIBA® gedurende meer dan 24 uur berust op de combinatie van twee mechanismen, waardoor trombinegeneratie verhoogd wordt: een toediening van geactiveerde stollingsfactoren (II, VII, IX, X) en tegelijkertijd een toename van het substraat voor emicizumab: FIX en FX (Hartmann, 2018).
Er zijn op dit moment onvoldoende data gepubliceerd en er is onvoldoende ervaring om een beslissing te maken over de noodzaak van remmer eradicatie in dit tijdperk van emicizumab. Belangrijke argumenten zullen zijn of behandeling met emicizumab ook na een periode van enkele jaren geen bijwerkingen heeft en of het aantal patiënten dat antistoffen tegen emicizumab maakt laag blijft. Derhalve is het op dit moment niet duidelijk of ITI achterwege gelaten kan worden. Zie voor verdere overwegingen de paper van Carcao et al. (Carcao, 2019).
Wat zijn de diagnostische en therapeutische opties bij pijn en beperkingen veroorzaakt door gewrichts- en spierbloedingen in het verleden bij patiënten met hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Gewrichtsbloedingen veroorzaken schade aan alle componenten van het gewricht en kunnen leiden tot (chronische) synovitis, kraakbeenschade, veranderingen aan het subchondrale bot en een gefibroseerd kapsel.
(Chronische) synovitis
Bij een gewrichtsbloeding wordt het gewricht blootgesteld aan allerlei schadelijke componenten in het bloed, zoals ijzer en ontstekingsmediatoren. Het synovium is verantwoordelijk voor het opruimen hiervan. Tijdens het opruimproces na een gewrichtsbloeding komt toxisch heem vrij dat kan neerslaan in het synovium in de vorm van hemosiderine (Roosendaal, 1998). Dit induceert een ontstekingsreactie en proliferatie van het synovium. Hierdoor neemt de zuurstofbehoefte van het synovium toe wat de afgifte stimuleert van groeifactoren waaronder ‘vascular-derived endothelial growth factor’ (VEGF) en daarmee de neoangiogenese (Acharya, 2011) Zo kan er een vicieuze cirkel ontstaan van bloeding – inflammatie en proliferatie – bloeding, doordat het verdikte en ontstoken synovium vatbaar is voor mechanische schade en het ontstaan van fragiele nieuwe bloedvaten. Wanneer deze cirkel niet doorbroken wordt en de klachten langer dan drie maanden blijven bestaan, is er sprake van een chronische synovitis.
Hemofilie-artropathie
Door recidiverende bloedingen ontstaat naast hypertrofie van de synoviale membraan (onherstelbare) schade aan het kraakbeen van het gewricht, hemofilie-artropathie. Deze schade wordt zowel veroorzaakt door de synoviale veranderingen en de bijkomende verhoogde productie van katabole enzymen en cytokinen (Roosendaal, 1998) als door directe effecten van bloed op kraakbeen (Christensen, 2018). Onderzoek suggereert dat beide pathologische processen parallel aan elkaar verlopen en elkaar beïnvloeden. Intra-articulair bloed veroorzaakt activatie van de chondrocyten met productie van kraakbeen-degenererende enzymen en bovendien apoptose van de chondrocyt waardoor de schade niet hersteld kan worden (Roosendaal, 1999; Hooiveld, 2003). Bovendien treedt er activatie op van de synoviale membraan door depositie van ijzer afkomstig van afbraak van de erytrocyten (Valentino, 2007). Hierdoor ontstaat een ontstekingsreactie met productie van katabole enzymen en cytokinen. Door hyperplasie en inflammatie van de synoviale membraan is er een verhoogde zuurstofbehoefte leidend tot vaatnieuwvorming (Acharya, 2011). Deze nieuwe fragiele vaten zijn kwetsbaarder en kunnen een vicieuze cirkel induceren van bloeding – schade – bloeding.
De ossale veranderingen die gezien worden in het kader van hemofilie-artropathie bestaan uit cystevorming, osteoporose, subchondrale sclerose, osteofytformatie, verbreding van de epifyse (indien bloedingen optreden voor het sluiten van de groeischijf) en gewrichtsspleetvernauwing met daarbij standsafwijkingen. De pathofysiologie van deze ossale afwijkingen is lastig te onderzoeken en daarmee is het tot op heden niet goed begrepen. Ditzelfde geldt voor de (mate van) fibrosering van het gewrichtskapsel. Van het ontstaan van osteoporose wordt aangenomen dat het een multifactorieel proces is, veroorzaakt door een afname in fysieke belasting, de artropathie zelf, een verlaagde BMI en een eventuele HIV en/of hepatitis C infectie en diens behandeling. Bovendien blijkt een bloeding zelf ook direct te leiden tot activatie van osteoclasten (Melchiorre, 2012; Lau, 2014).
Diagnostiek
Voor de diagnostiek van gewrichtsklachten bij hemofilie zijn de anamnese en het lichamelijk onderzoek de belangrijkste pijlers. (Chronische) synovitis kenmerkt zich door een (meestal) pijnloze zwelling van het gewricht, palpabele zwelling van het synovium zonder of met slechts een geringe bewegingsbeperking. Dit gaat vaak gepaard met atrofie van de omgevende spieren en een directe instabiliteit van het gewricht. Op de röntgenfoto worden aanvankelijk slechts geringe afwijkingen gezien, zoals een zwelling van de weke delen, ontkalking en vergroting van de epifysair schijven. Met een echo-doppler kan synoviale hypertrofie en toegenomen vascularisatie (doppler) vastgesteld worden. Met ‘magnetic resonance imaging’ (MRI) kunnen proliferatie van de synoviale membraan en neerslag van ijzerpigment worden vastgesteld, met toevoeging van contrastvloeistof kan inflammatie gedetecteerd worden (Querol, 2012; Lundin, 2012) Hemofilie-artropathie wordt op het niveau van het aangedane gewricht gekenmerkt door pijn, bewegingsbeperking in meerdere richtingen en mogelijk standsafwijking. Tijdens opvlammingen kan er ook sprake zijn van een synoviale zwelling van het gewricht, wat per abuis voor een bloeding kan worden aangezien (Timmer, 2015). Echo kan helpen bij de differentiatie tussen een bloeding en een opvlamming van hemofilie-artropathie (Timmer, 2015).
Met name als er meer gewrichten van één extremiteit en meer extremiteiten zijn aangedaan zal de patiënt een toenemende achteruitgang in activiteiten en participatie ervaren. De pijn is onder te verdelen in startpijn, pijn na (langdurige) belasting en nachtelijke pijn. De pijnklachten kunnen wisselend aanwezig zijn. Er is geen directe relatie tussen afwijkingen op de röntgenfoto en de klachten van het desbetreffende gewricht (Wallny, 2002a). De mate van degeneratie kan worden bepaald met behulp van de score volgens Pettersson (Wallny, 2002a).
Conventionele radiologie wordt gebruikt om de ontwikkeling en progressie van hemofilie-artropathie te inventariseren. De foto’s van de desbetreffende gewrichten worden beoordeeld op de aanwezigheid van osteoporose, epifyseale verbreding, irregulariteit van het subchondrale oppervlak, gewrichtsspleetvernauwing, subchondrale cystevorming, erosies, incongruentie en deformiteiten (Petterson, 1980). Dit zijn veranderingen die pas later in de ontwikkeling van artropathie optreden.
Voor het opsporen van vroege veranderingen in een gewricht of voor planning van een orthopedische ingreep kan gebruik gemaakt worden van MRI.
Conclusies
|
SORT grade |
Conclusie |
|
C |
De therapie bij chronische artropathie wordt geïndividualiseerd op basis van het klinisch beeld en de sociale context en niet (alleen) op de afwijkingen op de beeldvorming. (expert opinion/consensus) |
|
C |
Pijnbestrijding en actieve oefentherapie, gericht op functioneren, kunnen beperkingen van activiteiten verminderen en de participatie doen toenemen. (Strike, 2016; expert opinion/consensus) |
|
C |
Bij (chronische) synovitis dient dagelijks profylaxe te worden gegeven gedurende zes weken in combinatie met actieve oefentherapie. (Roosendaal, 1995; expert opinion) |
|
B |
Bij (chronische) synovitis is (radioactieve) synoviorthese aangewezen bij onvoldoende resultaat na 12 weken dagelijkse profylaxe. (Querol-Giner, 2017; Rodriguez-Merchan, 2014) |
|
C |
Bij een (chronische) synovitis die niet reageert op verhoogde profylaxe met stollingsfactorconcentraat kan ook een intra-articulaire injectie met corticosteroiden worden overwogen om de vicieuze cirkel te doorbreken. (Shupak, 1988; Fernández, 1997) |
|
C |
Bij (chronische) synovitis is (artroscopische) synovectomie aangewezen als er op de X-foto’s/MRI cysten te zien zijn. (expert opinion/consensus) |
|
C |
Alvorens operatief ingrijpen te overwegen dient een periode van functionele oefentherapie te worden. Bij enkelklachten dient een operatieve ingreep vooraf te worden gegaan door een schoenaanpassing en/of een proefkoker. (De Kleijn, 2006; De la Corte-Rodriguez, 2015) |
|
B |
Patiënten met een pijnlijke invaliderende artropathie die niet reageert op conservatieve behandeling, kunnen mogelijk in aanmerking komen voor gewricht vervangende operaties c.q. enkelartrodesen, ongeacht de leeftijd. Het streven hierbij is vermindering van pijn en verbetering van activiteiten en participatie. (Bluth, 2013; Moore, 2016; Marshall Brooks, 2011; Eckers, 2018; Wendt, 2011) |
|
C |
Orthopedische ingrepen bij hemofilie dienen altijd in een ziekenhuis plaats te vinden waaraan een hemofilie behandelcentrum is verbonden en door een orthopedisch chirurg met ervaring met hemofilie. (expert opinion/consensus) |
Samenvatting literatuur
Resultaten
Algemeen
Er zijn geen gerandomiseerde klinische trials over de orthopedische behandeling van hemofilie-artropathie gepubliceerd; publicaties zijn veelal case series, voornamelijk over synovitisbehandeling en knieprotheses. Hieronder worden de resultaten besproken van de meest recente relevante literatuur.
Oefentherapie/conservatieve therapie
Onderdeel van de synovitis behandeling is fysiotherapie. Kracht en stabiliteit kunnen echter pas worden opgebouwd wanneer het synovitis beeld is verbeterd. Oefentherapie is gericht op stabiliteit van het desbetreffend gewricht en wordt pas meer functioneel als voldoende stabiliteit bereikt is en met name, de zwelling wegblijft. Altijd is het evenwicht tussen de belastbaarheid en belasting het uitgangspunt van de benadering. In geval van artropathie kan oefentherapie nuttig zijn bij het verbeteren van het fysieke functioneren en de algehele conditie, waarbij handhaven van activiteiten en participatie het hoofddoel is. De belasting en belastbaarheid dienen op elkaar te worden afgestemd.
Strike et al toonden met behulp van een Cochrane review aan dat de meeste oefenprogramma’s verbetering gaven van een of meer klachten met name pijn, bewegingsuitslagen, kracht en loopvermogen (Strike, 2016). Hydrotherapie is mogelijk effectiever ten aanzien van reductie van pijnklachten dan droog oefenen. Functionele oefeningen lijken effectiever dan statische oefeningen ter bevordering van de spierkracht (Strike, 2016).
In een studie onder 13 patiënten met artropathie werd het effect van driemaal per week oefentherapie of sport op de gewrichtsuitslagen vergeleken met 36 patiënten die geen oefentherapie kregen of niet aan sport deden (Harris, 2006). De oefengroep toonde een significante verbetering van de bewegingsuitslagen ten opzichte van de niet geoefende groep (p=0,003). Zij suggereren dat verdere achteruitgang van een gewricht kan worden voorkomen door verbetering van de spierkracht, mede omdat hierdoor ook bloedingen worden voorkomen.
Vooral bij enkelklachten is aandacht voor schoeisel, gebruik van inlays en orthoses en zo nodig orthopedisch schoeisel van belang om klachten te verminderen en chirurgisch ingrijpen uit te stellen (De la Corte-Rodriguez, 2015). Bij een orthopedische ingreep, onafhankelijk van het gewricht dat wordt geopereerd, is pre- en post operatief fysiotherapie van essentieel belang. Het is onderdeel van een comprehensive care setting die als doel heeft om een zo goed mogelijk eindresultaat te bereiken (De Kleijn, 2006).
Radiosynovectomie
Querol-Giner et al evalueerden het effect van radiosynovectomie (RS) in 71 patiënten bij wie in totaal 174 procedures werden uitgevoerd in verband met recidiverende bloedingen: 90Y in knieën en 186Re in ellebogen enkels en schouders (Querol-Giner, 2017). RS resulteerde in een significante reductie van het aantal bloedingen van in totaal 582 voor de interventie naar 168 na de interventie, P < 0,001). Men constateerde echter een leeftijdsafhankelijk toename van de artropathie die niet werd beïnvloed door het al dan niet hebben ondergaan van een RS.
Rodriguez-Merchan evalueerde 443 gewrichten bij 345 patiënten die werden behandeld met RS. De gemiddelde leeftijd was 23,7 jaar (range 6-53), en gemiddelde follow-up 18,5 jaar (range: 6 maanden – 38 jaar). Er werden 1 tot 3 injecties gegeven met een interval van 6 maanden. Het aantal bloedingen nam af met 64%, de mate van synovitis met 31% en de gewrichtspijn met 69%. Röntgenologisch was er geen verbetering te zien. 28 patiënten ondergingen uiteindelijk een artroscopische synovectomie. Ze concluderen dat RS is een veilige en gemakkelijk uit te voeren methode is voor de behandeling van synovitis, maar dat bij ernstige synovitis meer dan één procedure noodzakelijk is (Rodriguez-Merchan, 2014).
Intra-articulaire injectie met corticosteroïden
De behandeling van synovitis door middel van intra-articulaire corticosteroidinjectie is beschreven in enkele case series. Shupak et al evalueerden het effect van intra-articulaire behandeling met methylprednisolon in 19 gewrichten met synovitis van 10 hemofilie patiënten (Shupak, 1988). Een subjectieve verbetering na 24 uur werd beschreven bij 79% en dit effect hield aan tot 8 weken in 58%. Ook nam het aantal bloedingen af in de 8 weken na injectie. Een case serie van 10 knieën beschreven door Rodriguez-Merchán liet een afdoende resultaat in 70% zien 1 jaar na injectie met methylprednisolon (Rodriguez-Merchán, 1994). Fernández-Palazzi et al evalueerden het effect van herhaalde injecties met dexamethason bij chronische synovitis in 34 patiënten (Fernández-Palazzi, 1997). De dexamethason werd herhaaldelijk toegediend, 3 maal à 3 weken waarna minimaal 6 maanden rust en dit kon tot 3 maal herhaald worden. Dit resulteerde in een goede subjectieve verbetering bij 19 patiënten en een goede objectieve verbetering bij 22 patiënten. Een recente studie van Martin et al beschrijft het effect van echogeleide corticosteroid injectie bij artropathische pijn in 25 patiënten (14 enkels, 13 ellebogen, 18 knieën) (Martin, 2017). De mediane pijnscore daalde van 7/10 naar 1/10, meestal al binnen 24 uur. De pijnreductie hield mediaan 8 weken aan (range 1-16 weken).
Osteotomie
De indicatie voor een correctieve osteotomie van femur of tibia is artropathie al dan niet in combinatie met een standsafwijking van het gewricht om een gewrichtsvervangende operatie te voorkomen. Gezien de goede resultaten van gewrichtsvervangende operaties (zie verder), wordt het echter in de praktijk niet vaak meer verricht, en heeft ook niet de voorkeur. Wallny et al verrichten bij 9 patiënten 11 intertrochantaire varus osteotomieen ivm artropathie van de heup. In zeven gevallen werd een klinische verbetering gezien, tweemaal een verslechtering en tweemaal geen verandering van het klinisch beeld. Ze stellen dat op basis van hun studie niet geconcludeerd kan worden dat een trochanter osteotomie altijd te verkiezen is boven een totale heupprothese (Wallny, 2002b).
Trieb et al rapporteerden de resultaten van 10 osteotomieën bij 7 patientën. Er werd zeven maal een osteotomie van de tibia en driemaal een supracondylaire osteotomie verricht. Zeven keer werd een verbetering van de klachten gezien, maar er trad geen verbetering van de knie functie op. Na gemiddeld 6,6 jaar werd bij vier patiënten zes maal een totale knie prothese geplaatst ivm toename van de klachten. Ze concluderen dat een osteotomie leidt tot uitstel van plaatsen van een knieprothese (Trieb, 2004).
Radiuskopextirpatie
De indicatie voor een radiuskopextirpatie is mechanische blokkade, pijn, synoviale impinchement en recidiverende bloedingen ten gevolge van erosie en vergroting van de radiuskop. Atalar et al rapporteerden de resultaten van radius extirpatie bij 14 patiënten met ernstige artropathie met pijn: een verbetering van pijn (VAS 6,5 naar 2), verbetering van rotaties en goed effect op het dagelijks functioneren. De VAS pijn score nam af van 6,5 naar 2,2, de rotatie van de elleboog verbeterde en alle 14 patiënten toonden een verbetering van het dagelijks functioneren (Mayo elbow performance score). Ze concluderen dat extirpatie van het radiuskopje een effectieve en veilige ingreep is voor patiënten met ernstige radiocapitellaire artropathie (Atalar, 2016). Silva et al evalueerden de resultaten van 40 extirpaties van de radius kop. Gemiddelde follow-up was 7,7 jaar. Na 5 jaar 63 graden verbetering van pro- en supinatie werd gezien, maar slechts 2 graden verbetering van de flexie en extensie. Er was een complicatie in de vorm van zenuw uitval die na 6 maanden was genezen. Zeven patiënten hadden een tweede ingreep nodig om het uiteindelijke resultaat te bereiken (Silva, 2007).
Enkel artrodese
De indicatie voor een enkelartrodese is pijn en dysfunctioneren als gevolg van ernstige artropathie. Bluth et al evalueerden de resultaten van 57 enkelartrodesen van het bovenste en/of onderste spronggewricht (BSG/OSG) bij 45 patiënten. Gemiddelde follow-up was 6,6 jaar. Aanvankelijk trad in 10% geen vergroeiing op na een artrodese van het BSG, en in 8.3% na een artrodese van het OSG. Na introductie van een nieuwe operatietechniek was dit 3,5 respectievelijk 5,6%. 75% van de patiënten had geen pijn meer, de overige 25% had een VAS score van 3-10. Patiënten functioneerden goed en loopafstand was sterk verbeterd. Zij concluderen dat artrodese de pijn vermindert en het functioneren verbetert (Bluth, 2013).
Knievervanging
De indicatie voor een knievervanging (TKP) is ernstige artropathie met pijn, en/of verminderd functioneren en/of recidiverende ernstige bloedingen.
Een meta-analyse van Moore et al onder 20 studies laat de resultaten zien van 336 TKPs bij 254 patiënten met hemofilieartropathie. De gemiddelde follow-up was 6,3 jaar. Er werd een significante verbetering van de knie functie gezien (10 gr extensie en 16 gr flexie van de knie), het effect op pijn wordt niet gemeld. Klinische en functionele verbetering gemeten middels knie-specifieke vragenlijsten was significant (klinisch 37,9 punten verbetering (effect size 3,21) en functioneel 13,5 punten toename (effect size 1,5). Na 106 operaties werd een complicatie gemeld. Het betrof vooral bloedingen (26x), late infecties (12x) en loslating (11x). In 21 gevallen was een revisie noodzakelijk. Ze concluderen dat TKP leidt tot een toename activiteiten van de patiënt (Moore, 2016).
Heupvervanging
De indicatie voor heup vervanging (THP) is pijn en functionele beperking. In een review evalueerden Parsa et al in 11 case reports en 9 series het effect van THP bij hemofilie patiënten. Zij concludeerden dat de resultaten, net als bij deze ingreep bij niet hemofilie, goed zijn mits de stolling goed wordt gecontroleerd en de revalidatie rustig wordt uitgevoerd (Parsa, 2018).
“Multiple-joint procedures”
Indien meer dan één gewricht ernstig is aangedaan kan overwogen worden om tijdens een opname meer dan een orthopedische ingreep te laten plaats vinden, zogenaamde multi-joint procedures. Dit spaart stollingsfactorconcentraten maar vereist een intensieve revalidatie en begeleiding door een ervaren team bestaande uit fysiotherapeut, revalidatie arts, orthopedisch chirurg, hemofiliearts en verpleegkundige (Schild, 2009). Gezien de belasting is begeleiding door de maatschappelijk werker van het hemofiliebehandelcentrum aangewezen. De Kleijn et al beschreven de ervaring van meer dan één orthopedische ingreep tijdens een opname bij 54 patiënten met een gemiddelde follow-up van 12 jaar. Er werden tien verschillende combinaties van THP, TKP en enkelartrodeses uitgevoerd. Het klinisch verloop was van deze groep was gunstig. Er een verbetering in activiteiten en participatie (De Kleijn, 2011).
Referenties
Zoeken en selecteren
Er werd voor deze uitgangsvraag in twee tempi een literatuuronderzoek verricht.
Voor de literatuur aangaande orthopedische ingrepen is een brede systematische review verricht. Er werd gezocht met de volgende zoektermen: haemophilia AND (arthropathy OR synovitis OR pseudotumor OR contracture) AND (treatment OR therapy) in PubMed zonder restricties. Dit resulteerde in 2050 artikelen. De volgende exclusie criteria werden toegepast: case reports, dierstudies, artikelen over de behandeling van hemofilie in het algemeen (i.e. PK studies, gentherapie, hemostase rondom orthopedische ingrepen etc.), artikelen over uitsluitend fysiotherapie / manuele therapie, pathofysiologie, diagnostiek, kwaliteit van leven bij artropathie van vóór 1990. Na screening van titel en abstract werden 9 artikelen geselecteerde. Op basis van de volledige tekst werden de meest relevante publicaties geselecteerd, deze staan weergegeven in de referentielijst.
Daarnaast is gebruik gemaakt van een Cochrane review uit 2016 gericht op beweeginterventies bij hemofilie (Strike, 2016). Een aanvullende zoekstring met zoektermen “haemophilia AND exercise” vanaf algemeen (niet als uitkomst van interventie), niet-hemofilie, specifieke ervaring lage lonen landen (behalve voor pseudotumor gezien beperkt voorkomen in landen met voldoende stolling), trombo-embolische complicaties, taal anders dan Engels/Frans/Duits, beschrijving van orthopedische status van inhibitorpatiënten (niet therapie), artikelen van vóór 1990. Na titel en abstract screening werden 242 publicaties geselecteerd. Vervolgens werden op basis van de volledige tekst de meest relevante publicaties (originele artikels, reviews en richtlijnen) geselecteerd. Deze staan weergegeven in de referentielijst. Er werd bovendien gebruik gemaakt van de richtlijnen van de WFH website (WFH Guidelines for the Management of Hemophilia, Joint Replacement Surgery in Hemophilia, Manufactured Shoes and Orthopedic Shoes).
Voor de literatuur aangaande fysiotherapie en manuele therapie is de volgende zoekstrategie gehanteerd: zoektermen “haemophilia AND manual therapy” resulteerde in 38 artikelen. De volgende exclusie criteria werden toegepast: dierstudies, behandeling anders dan manuele therapie, kwaliteit van leven bij artropathie algemeen (niet als uitkomst van interventie), klinimetrische studies, niet-hemofilie, taal anders dan Engels/Frans/Duits, artikelen december 2016 resulteerde in 72 artikelen. De volgende exclusie criteria werden toegepast: dierstudies, geen beweeginterventie, kwaliteit van leven bij artropathie algemeen (niet als uitkomst van interventie), klinimetrische studies, niet hemofilie artropathie of synovitis, taal anders dan Engels/Frans/Duits, artikelen van vóór 1990. Na screening van titel en abstract werden 9 publicaties geselecteerd. Op basis van de volledige tekst werden de meest relevante publicaties geselecteerd, deze staan weergegeven in de referentielijst.
Overwegingen
Ad aanbevelingen: Artropathie
Hemofilie-artropathie wordt gekenmerkt door pijn, bewegingsbeperking in meerdere richtingen en mogelijk standsafwijking. Vaak zijn meer dan één gewricht van één extremiteit en meer extremiteiten aangedaan. Met behulp van röntgenfoto’s kan de mate van degeneratie worden geobjectiveerd, hierbij wordt de score van Pettersson gebruikt (Pettersson, 1980). Lichamelijk onderzoek met behulp van de hemophilia joint health score (HJHS) en Hemophilia Activity list (HAL) zijn goede methodes om de functie en activiteit bij artropathie te evalueren (van Genderen, 2006; Hilliard, 2006)
De behandeling is in eerste instantie conservatief en gericht op pijnbestrijding in combinatie, leefstijladviezen verwijzing naar een fysiotherapeut en eventueel (schoen)aanpassingen, met als doel functionele verbetering en toename participatie.
Pijnbestrijding bij artropathie kan volgens onderstaand schema:
NB! Bij opiaten denk aan toevoegen van laxantia in het kader van obstipatie.
NSAID’s zijn gecontra-indiceerd i.v.m. het negatieve effect op de plaatjesfunctie. Een enkele keer wordt ibuprofen als pijnstiller gegeven:
Ook wordt ter verlichting van de pijn wel TENS (transcutane elektrische stimulatie) toegepast.
Indien chronische pijn niet met bovenstaande middelen te bestrijden is, bestaat er een indicatie voor orthopedisch ingrijpen of verwijzen naar het pijnteam (poli anesthesie).
Ad aanbevelingen: Synovitis
Conservatieve behandeling
Synovitis is een zwelling in een gewricht die onvoldoende reageert op toediening van stollingsfactoren, met meestal een normale bewegingsuitslag van het gewricht en geringe pijnklachten. Dit wordt vaak voorafgegaan door een toename van de bloedingsfrequentie in het aangedane gewricht of een (onder behandelde) ernstige bloeding. Het is een reactie van de synovia op bloed, die op termijn leidt tot kraakbeenschade en artropathie. Synovitis leidt tot instabiliteit, verlies van spierkracht, verminderde propriocepsis en geringer activiteiten- en participatieniveau. Chronische synovitis (synovitis die langer dan 6 weken bestaat) kan worden voorkomen door verder bloedingen te voorkomen.
De initiële behandeling van synovitis is gebaseerd op de volgende drie pijlers:
Follow-up
Indien na 6 weken een duidelijke verbetering is te zien kan terug worden gegaan naar standaard profylaxe. Fysiotherapie continueren indien nog spierfunctieverlies.
Indien na 6 weken geen verbetering, wordt het beleid gecontinueerd en vindt na 12 weken opnieuw evaluatie plaats. Indien dan nog geen verbetering is opgetreden, bestaat er een indicatie voor een synovectomie (radioactief dan wel artroscopisch).
Synovectomie
Wanneer na drie maanden behandeling geen verbetering optreedt, kan met behulp van echo of MRI de afwijkingen in het gewricht (onder andere synoviale membraan) geobjectiveerd worden. Een synoviorthese met radioactief Yttrium-90 of Rhenium-188 is geïndiceerd als er hypertrofie van de synoviale membraan en ijzer (hemosiderine)neerslag worden aangetoond (Querol-Giner, 2017). Er bestaat een contra-indicatie voor gebruik van radioactief materiaal indien de botcortex doorbroken is; dit in verband met het verslepen van radioactief materiaal buiten de intra-articulaire ruimte.
Bij synoviorthese wordt vier dagen substitutietherapie gegeven (dag 1: streef topspiegel 80-100 IE/dL dag 2-4: streef topspiegel 40-50 IE/dL. Het geïnjecteerde gewricht wordt gedurende drie dagen geïmmobiliseerd. Yttrium-90 en Rhenium-188 hebben een geringe penetratie en een korte halfwaardetijd en worden ook wel bij jonge kinderen toegepast (Rodriguez-Merchan, 2015a). De behandeling kan zo nodig, wanneer er na zes maanden onvoldoende effect is, worden herhaald (De La Corte-Rodriguez, 2013).
Wanneer op de MRI een forse hypertrofie van de synovia wordt vastgesteld, kan er een indicatie zijn voor een artroscopische of open synovectomie. Bij de eerste zijn de littekens kleiner, is de postoperatieve fase minder pijnlijk en zijn de actieve oefeningen gemakkelijker uit te voeren maar de kans op een postoperatief hematoom groter. In veel onderzoeken is een afname van de bloedingsfrequentie door deze procedure beschreven (Rodriguez-Merchan, 2014). Ook bij kinderen worden artroscopische synovectomieën succesvol uitgevoerd (Dunn, 2004 Journeycake 2003).
Na open synovectomieën heeft een hoger percentage patiënten een blijvende bewegingsbeperking van het gewricht. Beide technieken hebben een kans op residu dan wel recidief van de synovitis aangezien en dergelijke procedure nooit volledig is. Na elke synovectomie volgt een revalidatie traject. Na alle genoemde procedures is een begeleiding door een fysiotherapeut geïndiceerd om optimaal functioneel herstel na te streven (De Kleijn, 2006).
Intra-articulaire injectie met corticosteroïden
In Nederland worden corticosteroïden weinig toegepast in de context van hemofilie. De data over intra-articulaire behandeling met corticosteroïden bij synovitis zijn schaars. Subjectieve verbetering is beschreven. Men dient zich te realiseren dat het effect veelal kortdurend is en het een invasieve behandeling betreft waarvoor correctie van de stolling noodzakelijk is. In ervaren handen kan het overwogen worden om een vicieuze cirkel te doorbreken als adequate factor VIII of IX toediening onvoldoende resultaat heeft gegeven.
Ad aanbeveling: Chirurgische ingrepen
Het te verwachten resultaat na orthopedische ingrepen is met name vermindering van de pijn, waardoor meer functionele activiteiten en participatie mogelijk worden.
Nettoyage
Bij beginnende kraakbeenschade kunnen kraakbeen en botresten middels artroscopie worden verwijderd, zogenaamde nettoyage. Een nettoyage van de enkel kan uitkomst bieden wanneer conservatieve maatregelen niet meer afdoende zijn en er nog geen indicatie is voor artrodese, dan wel de wens dit uit te stellen (Rodriguez-Merchan, 2015b). Soms wordt dit gecombineerd met enkeldistractie in studieverband (zie verderop).
Osteotomie
Osteotomie is een standscorrectie in varus-/valgus- of flexie-/extensierichting, of een derotatie door middel van het doornemen en fixeren van bot. Dit kan een goede oplossing zijn voor bijvoorbeeld heup- en knieproblemen waarbij op bepaalde delen van het articulerende gewricht nog voldoende kraakbeen aanwezig is (Wallny, 2002b). Dit geldt met name als er contra-indicaties bestaan om een prothese te plaatsen, bijvoorbeeld bij jongere patiënten of patiënten met antistoffen tegen stollingsfactoren. Deze ingreep hoeft geen belemmering te vormen voor een eventueel later te plaatsen prothese (Trieb, 2004).
Extirpatie caput radii (radiuskopje)
Wanneer de pijn en de bewegingsbeperking in de elleboog worden veroorzaakt door een misvorming van het radiuskopje, kan een extirpatie in combinatie met synovectomie geïndiceerd zijn (Atalar, 2016; Silva 2007). Dit geeft vooral een verbetering van de pro- en supinatie en afname van de pijn. De stabiliteit van de elleboog wordt minder. Wanneer de patiënt de armen frequent gebruikt voor transfers (bijvoorbeeld opstaan uit een stoel), kan dit een relatieve contra-indicatie zijn.
Artrodese
Bij ernstige artropathie van het bovenste spronggewricht van de enkel, die gepaard gaat met een forse bewegingsbeperking, kan een enkelartrodese worden verricht (Bluth, 2013). De nabehandeling bestaat uit 12 weken gips, waarvan zes weken onbelast. De patiënt heeft na de artrodese minder pijn en kan met een afwikkelvoorziening onder de schoen een goed looppatroon bereiken (Lane, 2014). Soms is ook een artrodese van het onderste spronggewricht geïndiceerd. In een enkel geval vindt een pan-artrodese in een sessie plaats.
Gewricht vervangende operaties
Deze betreffen voornamelijk de knie en heup vervanging, en in veel mindere mate de elleboog, enkel en schouder (Moore, 2016; Marshall Brooks 2011; Eckers 2018; Wendt, 2011). De lange termijn resultaten wat betreft pijnvermindering en behoud van functionaliteit zijn goed. Bij een pijnlijke artropathie geven kunstgewrichten in ellebogen, knieën en heupen evenals een enkel artrodese vermindering van pijn en een toename van activiteiten en participatie. Belangrijkste doel van de ingreep is pijnbestrijding. Er worden ook enkelprothesen geplaatst bij patiënten met hemofilie, hoewel nog beperkt in aantal. Het voordeel ten opzichte van de artrodese is een betere functie van het enkelgewricht postoperatief, nadeel is de levensduur van de prothese en de frrequent waargenomen complicaties (Rodriguez-Merchan, 2015c).
Voor knie- en heupprotheses geldt dat hoewel er significant meer bloedingen optreden postoperatief, er geen verhoogd infectierisico is en de overlevingsduur van de protheses vergelijkbaar is met de niet-hemofilie populatie (Sikkema, 2011).
Er is ervaring met het combineren van meerdere ingrepen in één sessie, bijvoorbeeld twee knieprothesen. De totale revalidatieduur van de desbetreffende patiënt wordt hierdoor verkort. Bovendien is er minder stollingsfactorconcentraat nodig (Horoszowski, 1996; De Kliejn, 2014). De revalidatie na een dergelijke multiple-joint-ingreep vereist een ervaren team, dat minimaal bestaat uit een orthopedisch chirurg, hemofiliebehandelaar, fysiotherapeut en revalidatiearts.
Distractie
Distractie is het middels een fixateur externe (Ilazarov frame) uitéén rekken van de enkel, waardoor kraakbeen herstel kan optreden. In onderzoek verband wordt bij patiënten jonger dan 55 jaar met ernstige artropathie van het bovenste spronggewricht enkeldistractie met behulp van een fixateur externe verricht. De eerste korte termijnresultaten laten een gunstig effect op de klachten en behoud van functionaliteit zien (Van Vulpen, 2017). Er waren geen bloedingscomplicaties, wel had het merendeel van de patiënten een pengatinfectie, die in alle gevallen succesvol behandeld kon worden met een korte antibioticumkuur. Structurele veranderingen waren al na een jaar zichtbaar op röntgenfoto’s en MRI’s in de vorm van een afname van botcysten en botoedeem, en een verwijding van de gewrichtsspleet. Dit onderzoek is nog gaande, maar enkeldistractie lijkt een veelbelovend alternatief voor jonge hemofilie patiënten met ernstige enkelartropathie om een artrodese uit te stellen en de beweeglijkheid van het gewricht te behouden. Deze techniek is ook succesvol is gebleken voor knieartrose en kan dus mogelijk ook voor knieartropathie worden toegepast (Van der Woude, 2017).
De hemofilie populatie wordt ouder. De levensverwachting benadert die van de algemene mannelijke populatie. Met de toenemende leeftijd ontstaan leeftijdsgebonden comorbiditeiten. In patiënten met hemofilie zal het behandelteam in het hemofiliebehandelcentrum een actieve rol moeten nemen in coördinatie en begeleiding binnen bestaande preventieve programma’s zoals screening op colorectaal carcinoom en screenen op cardiovasculaire risicofactoren. Bij het uitvoeren van aanvullend onderzoek of voorschrijven van medicatie is het belangrijk dat kennis over hemofilie(behandeling) wordt meegewogen.
Aanbevelingen
Onderbouwing
Inleiding
In dit onderdeel is onderzocht in hoeverre cardiovasculaire risico factoren voorkomen in patiënten met hemofilie in vergelijking met de algemene populatie. Het onderzoek richt zich op hypertensie, hyperlipidemie, roken, obesitas en diabetes mellitus. Het achterliggende doel is om gegronde adviezen te geven over de noodzaak voor screening op cardiovasculaire risicofactoren in hemofilie patiënten.
In tabel 11.1.2 is de ‘Summary of Findings’ tabel opgesteld. In deze tabel staan de uitkomstmaten centraal, met andere woorden de cardiovasculaire risicofactoren staan in de eerste kolom. Alle studies waarin de vergelijking van prevalenties van cardiovasculaire risico factoren tussen levende, volwassen hemofilie patiënten en de algemene populatie centraal staan, worden meegenomen in de tabel. De resultaten van de verschillende studies zijn per uitkomst opgesomd. De data zijn niet gepoold, omdat de studies onderling te veel verschillen qua leeftijd, populaties waaruit samples hemofilie patiënten komen, vergelijkende populaties en definities. Alle studies in de ‘Summary of Findings’ tabel zijn observationeel.
Conclusies
|
SORT Grade |
Conclusie |
|
B |
Hypertensie wordt vaker bij hemofilie patiënten gezien dan in de algemene populatie. (Biere-Rafi, 2011; Sharathkumar, 2011; Fransen van de Putte, 2013; Drygalski, 2013; Pocoski, 2014; Sait, 2014; Wang, 2015) |
|
B |
Hypercholesterolemie wordt minder vaak bij hemofilie patiënten gezien dan in de algemene populatie. (Rosendaal, 1990; Biere-Rafi, 2011; Sharathkumar, 2011; Fransen van de Putte, 2013; Pocoski, 2014; Sait, 2014; Wang, 2015; Sood, 2018) |
|
B |
Patiënten met hemofilie lijken minder vaak te roken dan mannen in de algemene populatie. (Sharathkumar, 2011; Lim, 2011; Fransen van de Putte, 2013; Sait, 2014; Barnes, 2016; Sood, 2018) |
|
B |
Er lijkt geen verschil te zijn in de prevalentie van obesitas tussen hemofilie patiënten en de algemene populatie. (Hofstede, 2008; Biere-Rafi, 2011; Sharathkumar, 2011; Lim, 2011; Fransen van de Putte, 2013; Wang, 2015; Barnes, 2016; Sood, 2018) |
|
B |
Er lijkt geen verschil te zijn in de prevalentie van diabetes mellitus tussen hemofilie patiënten en de algemene populatie. (Sharathkumar, 2011;Lim 2011; Fransen van de Putte, 2013; Wang, 2015; Sood, 2018) |
Samenvatting literatuur
Resultaten
De gevonden publicaties zijn observationeel. Sommigen zijn cross-sectioneel van opzet, anderen retrospectief. Twee studies hebben een prospectieve opzet (Soucie, 2011; Curtis, 2015). Een aantal studies heeft als doel het vaststellen van de prevalentie van cardiovasculaire risico factoren in patiënten met hemofilie in vergelijking met de algemene mannelijke populatie (bijv. Rosendaal, 1990; Biere-Rafi, 2011; Khleif, 2011; Fransen van de Putte, 2012). In andere studies wordt alleen de prevalentie van cardiovasculaire risico factoren vastgesteld in een cohort hemofilie patiënten (Minuk, 2015; Sartori, 2008), wordt de relatie tussen cardiovasculaire risico factoren en cardiovasculaire incidenten in hemofilie patiënten onderzocht (Kulkarni, 2005), of wordt er een vergelijking gemaakt tussen de incidentie van cardiovasculaire aandoeningen bij hemofilie patiënten en de algemene populatie (Kulkarni, 2005; Fransen van der Putte, 2012). In een aantal studies is kwaliteit van leven het primaire onderzoeksdoel (Siboni, 2009; Von Mackensen, 2012). De leeftijden van de geïncludeerde hemofilie patiënten verschillen per studie. Sommige studies onderzoeken een cohort hemofilie patiënten vanaf 18 jaar (Biere-Rafi, 2011; Drygalski, 2013), andere studies vanaf 30, 40 of 65 jaar (Fransen van de Putte, 2012; Khleif, 2011; Berger, 2016; Von Mackensen, 2012). Er zijn ook studies naar kinderen en jong volwassenen (Revel-Vilk, 2011; Alperstein, 2018; Limjoco, 2018). Ook de vergelijkende populaties verschillen per studie. Vaak worden er gegevens gebruikt uit grote surveys onder de algemene bevolking (Rosendaal, 1990; Sharathkumar, 2011; Lim 2011), soms wordt er heel specifiek gematched (Sartori, 2008) en in andere studies worden patiënten zonder hemofilie gebruikt als vergelijkende populatie (Siboni, 2009; Ragni, 2011).
Alle studies waarin de vergelijking tussen de prevalenties van cardiovasculaire risico factoren tussen, volwassen hemofilie patiënten en de algemene populatie centraal staat, worden meegenomen in Tabel 11.1.2. In deze ‘Summary of Findings Table’ worden de resultaten van de verschillende studies per uitkomst opgesomd. Alle studies die gebruikt zijn in deze hebben een studie kwaliteit van level 2 volgens SORT (limited quality patient-oriented evidence). Er zijn overigens geen data gevonden in hoeverre de behandeling van hypertensie in patiënten met hemofilie verschilt met die in patiënten zonder hemofilie.
Hypertensie
Uit de gevonden studies blijkt dat hypertensie vaker voorkomt in patiënten met hemofilie dan in de algemene populatie. De oorzaak hiervan is nog niet opgehelderd. In de studie van Sun et al (Sun, 2016) was hypertensie niet geassocieerd met hematurie. Dit werd ook gezien in de resultaten uit de Europese Advance groep (Holme, 2016). Daarnaast bleken, net als in de algemene populatie, leeftijd en Body Mass Index (BMI) van invloed op hypertensie bij patiënten met hemofilie (Holme, 2016). Renale dysfunctie was geassocieerd met hypertensie in de Advance en de ARIC studies (Holme, 2016; Sood, 2018), terwijl dit niet werd gezien door anderen (Fransen van de Putte, 2013). Vanuit een ander oogpunt is gesuggereerd dat vasculaire gewrichts ‘remodelling’ geassocieerd is met hypertensie (Barnes, 2017).
Hypercholesterolemie
De meeste studies laten zien dat hypercholesterolemie minder vaak voorkomt in hemofilie patiënten in vergelijking met de algemene populatie. De gemiddelde totale cholesterol levels en de totaal/HDL ratio’s waren lager in patiënten met een actieve chronische hepatitis C infectie (Fransen van de Putte, 2013). Een specifieke maatregel voor hemofilie wordt dan ook niet ondersteund.
Overgewicht en obesitas
De toenemende prevalentie van overgewicht en obesitas in de algemene bevolking is ook zichtbaar in de hemofilie populatie. Dit geldt zowel voor kinderen als volwassenen, voor ernstige en niet-ernstige hemofilie. Behalve de cardiovasculaire implicaties, heeft obesitas ook hemofilie-specifieke complicaties. De bewegingsbeperking neemt toe met de BMI (Soucie, 2004), de kans op heupafwijkingen stijgt van 16 naar 24% (Kelly, 2013), er is meer schade aan de gewrichten (Carpenter, 2006) en de kans op succesvolle zelf-infusie vermindert (Ullmann, 2014). In de obese niet-hemofilie populatie gaat gewichtsverlies gepaard met minder gewrichtsklachten en een betere functie. Als laatste heeft overgewicht een flinke impact op de dosis stollingsfactoren die wordt voorgeschreven.
Referenties
Bewijskracht literatuur
De studies die gebruikt zijn hebben een studie kwaliteit van level 2 volgens SORT.
Zoekverantwoording
De verschillende onderdelen van de zoekstrategie (zie Tabel 11.1.1) zijn gecombineerd tot een uiteindelijke zoekopdracht in PubMed: (((((Hemophilia A[MeSH Terms]) OR Hemophilia B[MeSH Terms]) OR hemophilia[Title/Abstract]) OR haemophilia[Title/Abstract])) AND (((((cardiovascular[Title/Abstract]) AND risk factor*[Title/Abstract])) OR ((“Hypertension”[Mesh]) OR ((hypertension[Title/Abstract]) OR blood pressure[Title/Abstract]))) OR ((((obesity[Title/Abstract]) OR hypercholesterolemia[Title/Abstract]) OR diabetes mellitus[Title/Abstract]) OR smoking[Title/Abstract])).
Tabel 11.1.1. Opbouw zoekstrategie PubMed
|
Keyword |
MeSH database |
Title and Abstract (tiab) |
Combineren (MesH/tiab) |
|
Hemophilia |
(“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh]): 21523
|
((hemophilia[Title/Abstract]) OR haemophilia[Title/Abstract]): 18985
|
(((Hemophilia A[MeSH Terms]) OR Hemophilia B[MeSH Terms]) OR hemophilia[Title/Abstract]) OR haemophilia[Title/Abstract]): 25427 |
|
Cardiovascular risk factors |
“Risk Factors”[Mesh] à onderaan de boom. Niet nuttig. |
(cardiovascular[Title/Abstract]) AND risk factor*[Title/Abstract]): 85472 |
|
|
Hypertension |
(“Hypertension”[Mesh]): 242174
|
((hypertension[Title/Abstract]) OR blood pressure[Title/Abstract]): 549444
|
(“Hypertension”[Mesh]) OR ((hypertension[Title/Abstract]) OR blood pressure[Title/Abstract]): 603981 |
|
‘Obesity’, ‘Hypercholesterolemia’, ‘Diabetes mellitus’, ‘Smoking’ |
|
(((obesity[Title/Abstract]) OR hypercholesterolemia [Title/Abstract]) OR diabetes mellitus [Title/Abstract]) OR smoking[Title/Abstract]: 585541 |
|
De zoekopdracht werd gedaan op 21 december 2018 en resulteerde in 302 publicaties. De resultaten van de zoekstrategie zijn gescreend op titel en samenvatting. Na deze eerste screening blijven er 59 publicaties over die mogelijk interessant zijn voor de uitgangsvragen. Na het controleren van de referenties blijken 4 additionele publicaties mogelijk van belang te zijn voor de uitgangsvragen niet gevonden met de initiële zoekstrategie in PubMed. In totaal zijn er daarmee 63 artikelen mogelijk interessant.
Wanneer aan de initiële zoekstrategie ‘AND (treatment OR prevention)’ toevoegd wordt, levert dit 127 resultaten op. Bij een vergelijking tussen de 302 initiële resultaten en de 127 resultaten na de laatste zoekstrategie, zijn er geen artikelen die bij de initiële zoekstrategie niet gevonden zijn.
Om gerichter naar studies te zoeken over de behandeling van hypertensie bij patiënten met hemofilie is de volgende zoekstrategie uitgevoerd: ((((((“hemophilia a”[MeSH Terms]) OR b, hemophilia[MeSH Terms]) OR hemophilia[Title/Abstract]) OR haemophilia[Title/Abstract])) AND ((((hypertension[MeSH Terms]) OR hypertension[Title/Abstract]) OR blood pressure[Title/Abstract]) OR antihypertensive[Title/Abstract])) AND ((prevention[Title/Abstract]) OR treatment[Title/Abstract]). Tussen de 73 resultaten zijn er geen artikelen die bij de initiële zoekstrategie niet gevonden zijn.
De Cochrane Database of Systematic Reviews bevat geen artikelen over het onderwerp cardiovasculaire risico factoren bij patiënten met hemofilie.
Tabel 11.1.2 Summary of Findings Table (pdf)
Overwegingen
Algemeen
Het doel van de NHG-standaard ‘Het PreventieConsult is de preventie van hart-en vaatziekten, diabetes mellitus type 2 en chronische nierschade door actief aanbod van een risicoschatting gekoppeld aan bijpassende adviezen of therapie. Omdat het hemofilie behandelcentrum een centrale plaats inneemt bij de behandeling van patiënten met hemofilie, is het van belang dat de hemofiliebehandelaar een actieve rol speelt in het cardiovasculair risicomanagement.
Er zijn geen studies gedaan naar het effect van cardiovasculaire risicoprogramma’s op het voorkómen van cardiovasculaire events bij hemofilie. Wel is aangetoond dat het cardiovasculaire risicoprofiel in hemofilie patiënten ongunstig is (Fransen van de Putte, 2013). Het is aannemelijk dat de preventieve werking van behandeling van risico factoren voor hart- en vaatziekten ook in de hemofilie populatie zichtbaar zal zijn in het voorkomen van hart- en vaatziekten. Juist vanwege de delicate balans tussen het trombose- en bloedingsrisico in patiënten met hemofilie die behandeld moeten worden met anticoagulantia, is het van belang om risico factoren vroegtijdig op te sporen en te behandelen. Er zijn meerdere publicaties (expert opinions) die cardiovasculaire risicoprogramma’s in hemofilie stimuleren (NVHB richtlijn, 2009; Mauser-Bunschoten, 2009; Dolan, 2009; Wong, 2011; Fransen van de Putte, 2012; Fransen van de Putte, 2013; Adams, 2014; Hermans, 2014; Kulkarni, 2014; Kamphuisen, 2014; Ullman, 2014; Sousos, 2016; Witkop, 2016; Kahan, 2017; Schutgens, 2017; Wilding, 2018).
Welke beleid voor chronische hepatitis C wordt bij patiënten met hemofilie geadviseerd?
Aanbevelingen
Onderbouwing
Inleiding
Hepatitis C besmetting
Bloedingen bij hemofilie patiënten worden voorkomen of behandeld door (profylactische) toediening van stollingsfactorconcentraten. Tussen 1979 en 1985 zijn 16% van de hemofilie patiënten besmet geraakt met HIV (human immunodeficiency virus). Tot 1992 is de meerderheid van patiënten met HCV (hepatitis C virus) als gevolg van toediening van besmette stollingsproducten geproduceerd uit humaan bloed. Patiënten die regelmatig zijn behandeld met stollingsfactorconcentraten vervaardigd uit grote plasmapools, zijn in vrijwel alle gevallen besmet met HCV. Bij patiënten die zijn behandeld met cryoprecipitaat is dit 66% (Mauser-Bunschoten, 1995; NVHB richtlijn, 2009). In Nederland zijn uit plasma-bereide stollingsfactorconcentraten, door adequate virusinactivatie, sinds 1985 vrij van HIV en sinds 1992 van HCV (Mauser-Bunschoten, 1995; Plug, 2006).
Chronische HCV infectie
Indien HCV-antistoffen positief zijn, dient onderzoek naar HCV-RNA te worden gedaan. Indien het HCV-RNA 2 keer positief is met een tussenpoos van 6 maanden, is er sprake van chronische hepatitis C (richtlijn NVHB, 2009). Van de personen die besmet zijn met HCV, ontwikkelt ongeveer 80% een chronische HCV infectie (Yee, 2000; Seeff, 2002; Posthouwer, 2007). Het natuurlijk beloop van de ziekte is variabel. Gedurende enkele tientallen jaren kan de HCV infectie stabiel zijn met weinig tot geen lever schade. Echter, op den duur kan een onbehandelde chronische HCV infectie resulteren in fibrose en cirrose, wat uiteindelijk kan leiden tot eindstadium leverziekte met gedecompenseerde levercirrose, varices bloedingen, hepatocellulair carcinoom en overlijden (Yee, 2000; Seeff, 2002). Tevens kan chronische HCV infectie leiden tot extra-hepatische manifestaties zoals auto-immuun- en lymfoproliferatieve ziekten (Negro, 2015). Uit een retrospectief onderzoek onder 863 patiënten met HCV en een erfelijke stollingsstoornis uit Nederland en Engeland bleek dat 19% van de patiënten het virus spontaan had geklaard en dat 81% een chronische HCV infectie ontwikkelde (Fransen van de Putte, 2014). De mediane follow-up sinds de HCV infectie was 31 jaar in dit cohort. Tijdens deze follow-up ontwikkelde 13% van de patiënten met chronische HCV eindstadium leverziekte. Hepatocellulair carcinoom werd in 3% van de patiënten gediagnosticeerd, waarvan 41% in de laatste 6 jaren van de follow-up. Gelijktijdige HIV infectie versnelt de progressie tot eindstadium leverziekte (Seeff, 2002; Fransen van de Putte, 2014).
Behandeling chronische HCV infectie
Het doel van de antivirale behandeling van hepatitis C is eradicatie van het virus om zo de complicaties van HCV-gerelateerde lever- en extra-hepatische aandoeningen te voorkomen (EASL guideline, 2018; Shapiro, 2018). Lange tijd (zie ook de vorige hemofilie-behandelrichtlijn uit 2009) bestond deze behandeling uit een combinatie van peginterferon en rivabarine gedurende 24 tot 48 weken. Deze behandeling was succesvol (‘sustained virological remission’ (SVR)) in ongeveer 50% van de patiënten met HCV genotype 1 of 4 en in 80-90% van de patiënten met genotype 2 of 3 (Manns 2001; Fried, 2002; Posthouwer, 2007). De behandeling met peginterferon en ribavirine wordt echter als zeer ingrijpend ervaren en gaat gepaard met veel bijwerkingen: o.a. griepachtig beeld, beenmergdepressie en psychische bijwerkingen (Farmacotherapeutisch Kompas, van Frans vd Putte, 2009 en 2011).
Vanaf eind 2014 zijn nieuwe direct werkende antivirale middelen (‘direct-acting antivirals’ (DAA’s)) beschikbaar in Nederland waardoor een volledige orale en interferon vrije behandeling mogelijk is geworden. Dit geldt voor HCV-geïnfecteerde patiënten die niet eerder zijn behandeld (initiële therapie) en voor patiënten waarbij een eerdere therapie heeft gefaald.
In juli 2018 is het ‘Richtsnoer behandeling hepatitis C infectie’ ge-update. Dit richtsnoer is opgesteld door een commissie bestaande uit verschillende beroepsverenigingen (NIV, NVHB, NVMDL, NVH, NVZA) en richtsnoer volgt de aanbevelingen van de ‘European Association for the Study of the Liver (EASL)’ guideline (EASL guideline, 2018). Omdat hepatitis C in hemofilie patiënten niet wezenlijk anders wordt behandeld dan in patiënten zonder hemofilie worden de twee richtlijnen als leidraad genomen voor de huidige aanbevelingen, en is er geen nieuwe literatuur zoekopdracht uitgevoerd naar dit onderwerp.
Conclusies
|
SORT Grade |
Conclusie |
|
A |
Progressie naar eindstadium leverziekte bij een chronische HCV infectie verschilt niet tussen patiënten met hemofilie en de algemene populatie. (EASL guideline, 2018) |
|
A |
De indicaties voor HCV therapie zijn dezelfde in patiënten met en zonder stollingsstoornis. (EASL guideline, 2018) |
|
B |
De interferon- en ribavirine-vrije anti-HCV behandelingen (HCV – Direct Acting Antivirals) die kunnen worden gebruikt in patiënten met een stollingsstoornis zijn dezelfde als in patiënten zonder stollingsstoornis. (Hezode 2017; EASL guideline, 2018) |
|
A |
Onbehandelde patiënten met chronische hepatitis C en degenen bij wie behandelingen gefaald hebben, moeten regelmatig vervolgd worden. (EASL guideline, 2018) |
|
A |
Niet-invasieve methoden voor stagering van fibrose zijn het meest geschikt voor evaluatie van chronische hepatitis C, met een interval van 1 tot 2 jaar (EASL guideline, 2018) |
Samenvatting literatuur
Resultaten
Behandeling van HCV
Het Farmacotherapeutisch Kompas verwijst voor informatie over de meest recente behandeladviezen naar het ‘Richtsnoer behandeling hepatitis C infectie’ uit juli 2018. Dit richtsnoer is een update van de eerder gepubliceerde richtlijnen betreffende hepatitis C (NVMDL richtlijn, 2011; Berden, 2014) en is opgesteld door een commissie bestaande uit verschillende beroepsverenigingen (NIV, NVHB, NVMDL, NVH, NVZA). Het richtsnoer volgt de aanbevelingen van de ‘European Association for the Study of the Liver’ (EASL guideline, 2018). Behandeling met een combinatie van DAA’s is zeer effectief en in 90-100% van de gevallen wordt een sustained virological response (SVR) bereikt. De behandeling is 8-12 weken) en brengt geen interferon-gerelateerde bijwerkingen met zich mee (Walsh, 2017; EASL guideline, 2018; Richtsnoer behandeling hepatitis C infectie, juli 2018).
Verschillende studies naar de behandeling met DAA’s in hemofilie patiënten laten vergelijkbare resultaten zien met de algemene populatie (Walsh 2017; Hézode, 2017). In een studie waarin HCV-positieve patiënten met de ziekte van Von Willebrand of hemofilie behandeld werden met grazoprevir en elbasvir (gedurende 12 weken, zonder ribavarine), was de SVR 91% (Hézode, 2017). De commissie die het ‘Richtsnoer behandeling hepatitis C infectie’ heeft opgesteld is van mening dat er een indicatie voor behandeling is met DAA’s voor iedere HCV patiënt in Nederland. Shapiro et al. (Shapiro, 2018) doet, in een recente review over de ouder wordende hemofilie patiënt, de volgende aanbeveling betreffende de behandeling met DAA’s: ‘offer all patients with hemophilia who have active hepatitis treatment to clear HCV with the newer agents as appropriate’. Dit werd bekrachtigd door een editorial (Makris, 2017).
De EASL richtlijn wijdt ook een hoofdstuk aan hemoglobinopathieën en stollingsstoornissen. Hierin is een aantal conclusies beschreven:
Follow-up van patiënten na eradicatie/SVR van chronische HCV infectie
In het ‘Richtsnoer behandeling hepatitis C infectie’ (juli 2018) wordt m.b.t. monitoring tijdens en follow-up na behandeling verwezen naar de EASL richtlijn (2018), omdat er nog te weinig literatuur is om een concreet advies hierover te geven. In de EASL richtlijn wordt een hoofdstuk gewijd aan de follow-up van patiënten die na behandeling een SVR bereiken. Voor patienten met gevorderde fibrose (F3) of cirrose (F4) wordt –na bereiken van SVR- echografie van de lever ter controle van ontwikkeling hepatocellulair carcinoom aanbevolen.
Follow-up van patiënten met chronische HCV infectie
In de EASL richtlijn wordt een kort hoofdstuk gewijd aan de follow-up van onbehandelde patiënten en van patiënten waarbij de behandeling heeft gefaald: deze patiënten moeten regelmatig onderzocht worden. De redenen voor de non-behandeling of het falen van behandeling dienen goed gedocumenteerd te worden.
Referenties
Bewijskracht literatuur
Level 1, consistent.
Zoekverantwoording
N.v.t.
Evidence tabellen
N.v.t.
Overwegingen
Besmetting met HCV komt nog voor bij patiënten die vóór 1992 zijn behandeld met besmette stollingsproducten geproduceerd uit humaan bloed. In Nederland is het streven om iedere patiënt met HCV besmetting te behandelen. De NVHB is van mening dat deze behandeling door maag-darm-leverartsen in nauwe samenwerking met hemofiliebehandelaars moet plaatsvinden. De aanbevelingen die zijn gebaseerd op de EASL guideline vinden oorsprong in een relatief kleine paragraaf (“Haemoglobinopathies and bleeding disorders”) onder “Treatment of special groups”, met als aanbeveling dat eenzelfde behandelindicatie voor deze groepen geldt als voor alle patiënten met HCV infectie.
Moeten hemofilie patiënten deelnemen aan het bevolkingsonderzoek darmkanker (RIVM)?
Aanbevelingen
Onderbouwing
Inleiding
In deze paragraaf onderzoeken we wat de waarde is van de (i)FOBT ((immunologisch) fecaal occult bloed test) voor het opsporen van coloncarcinoom bij patiënten met hemofilie. Is deze test vaker vals positief? En wat is bekend over de prevalentie van gastro-intestinale bloedingen en het uitvoeren van colonoscopie bij patiënten met hemofilie? Het onderzoek richt zich op deze uitgangsvragen, echter omdat over de (i)FOBT bij hemofilie patiënten geen literatuur beschikbaar bleek te zijn, is een aantal deelvragen geformuleerd en is ook de parallel gezocht met de waarde van de (i)FOBT bij patiënten die antistollingsmedicatie gebruiken. Voor de conclusies en aanbevelingen van het onderzoek is de sterkte van bewijsvoering laag en deels uitsluitend “expert opinion”.
Conclusies
|
SORT Grade |
Conclusie |
|
B |
Het lijkt aannemelijk dat patiënten met hemofilie eenzelfde prevalentie van colorectaal carcinoom hebben in vergelijking met de algemene populatie. (Fransen van de Putte, 2012; Huang, 2015) |
|
C |
Er zijn geen gegevens over de “positivity rate” van de (i)FOBT bij patiënten met hemofilie; bij patiënten die antistolling gebruiken is de “positivity rate” hoger dan in de algemene bevolking zonder toename van vals positieve uitslagen. (Nieuwenburg, 2019) |
|
B |
Een colonoscopie kan bij patiënten met hemofilie veilig worden uitgevoerd na volledige correctie van de stollingsfactor deficiëntie pre- en eventueel post-procedureel. (Tintillier, 2013; Fogarty, 2010; Tomaszewski, 2019) |
|
C |
Patiënten met hemofilie dienen een colonoscopie bij voorkeur te ondergaan in een hemofiliebehandelcentrum. (Fogarty, 2010; van Os, 1999) |
Samenvatting literatuur
Resultaten
De zoekvraag over de waarde van de (i)FOBT voor het opsporen van coloncarcinoom bij patiënten met hemofilie levert geen bruikbare literatuurverwijzingen op. Er zijn geen originele studies gedaan naar de waarde van de (i)FOBT als screeningsmethode bij het opsporen van coloncarcinoom bij patiënten met hemofilie. De ‘Malignancy in Haemophilia Workshop Group’ (Astermark, 2012) bespreekt helder de problemen en dillema’s rondom maligniteiten bij hemofilie patiënten. Zestien hemofiliebehandelaren participeerden in een “post-meeting survey”, waaruit de volgende twee conclusies met betrekking tot (i)FOBT worden beschreven (SORT level 3): 1. het is aannemelijk dat de (i)FOBT vaker positief is in de hemofilie populatie, echter de kans op aanwezigheid van colorectale maligniteit verschilt waarschijnlijk niet van de algemene populatie. 2. 94% zou altijd een colonoscopie adviseren bij een positieve (i)FOBT bij patiënten met hemofilie (ongeachte de ernst), als de oorzaak van de bloeding niet duidelijk is.
Coloncarcinoom bij patiënten met hemofilie
Fransen van de Putte et al. (Fransen van de Putte, 2012) onderzocht in 2012 retrospectief de cumulatieve incidentie van verschillende maligniteiten in een cohort hemofilie patiënten geboren voor 1971 in Nederland en vergeleek deze met de algemene Nederlandse mannelijke populatie. Er werd geen verschil gezien in het voorkomen van maligniteiten, ook niet van colorectaal carcinoom. De prevalentie van coloncarcinoom in de hemofilie patiënten (n=408) was 2,0% (95% CI 0,9-3,8) (n=8) versus 1,3% (95% CI 1,19-1,33) in leeftijd-gematchte algemene populatie. Een uitzondering is hepatocellulair carcinoom, vanwege de associatie met hepatitis C infectie. Huang et al. (Huang, 2015) beschreef retrospectief de cumulatieve incidentie van maligne ziekten bij 1054 hemofilipatienten uit een Taiwanees ziekenhuis en vond voor colorectaal carcinoom ook geen verschil met de algemene populatie. Op basis van deze beperkte data lijkt het onwaarschijnlijk dat er een verschil is in het voorkomen van coloncarcinoom tussen hemofilie patiënten en de algemene populatie. Bewijskracht literatuur: study quality level 2 (SORT).
Gastro-intestinale bloedingen in patiënten met hemofilie
Op basis van 12 artikelen resulterend uit de zoekopdracht naar het voorkomen van (occulte) colorectale bloedingen bij hemofilie patiënten (in vergelijking met de algemene populatie) kunnen geen conclusies getrokken worden. Eyster et al. rapporteren een incidentie van hoge GI-bloeding van 1,3% per jaar; dit is 10x hoger dan in de algemene bevolking (Eyster, 2007; Lanas, 2005). Spontane hemorragie van de darmwand zou voorkomen bij 4,85% van de ernstige hemofilie patiënten (Mittal, 1985). Pickles et al. 2018 vat samen: studies hebben getoond dat gastro-intestinale bloeding meestal wordt veroorzaakt door gastro-intestinale pathologie zoals maag-darm ulcera (met of zonder H. Pylori infectie) (Lee, 1997; Braden, 1998). De aanwezigheid van een stolfactor deficiëntie is dan niet dé oorzaak van de bloeding en adequaat onderzoek en behandeling moeten niet vertraagd worden ingezet. (Pickles, 2018). Bewijskracht literatuur: study quality level 3 (SORT).
Wat is de invloed van antistolling op de (i)FOBT
Uit de meta-analyse van Nieuwenburg et al. kan worden geconcludeerd dat de nauwkeurigheid van de (i)FOBT bij het opsporen van ‘advanced neoplasia’ en colorectaal carcinoom (“accuracy rate”) vergelijkbaar is tussen patiënten zonder en met orale anticoagulantia of NSAID’s. Op basis hiervan wordt het stoppen van orale anticoagulantia of NSAID’s voor (i)FOBT screening niet aanbevolen (Nieuwenburg, 2018). De Klerk et al. concluderen in hun meta-analyse dat gebruik van aspirine of anticoagulantia geen toename veroorzaken in het aantal vals-positieve test uitslagen (de Klerk, 2018). Een hogere ‘positivity rate’ in patiënten met orale anticoagulantia of NSAID’s (Bujanda, 2013) zou veroorzaakt kunnen worden door stimulatie van bloedingen uit laesies in het colon (zowel benigne als (pre)maligne) (Nieuwenburg, 2018). Daarnaast blijkt de positief voorspellende waarde van de (i)FOBT in personen met en zonder antitrombotische medicijnen vergelijkbaar (en voor DAPT-gebruikers zelfs hoger). Dit zou kunnen betekenen dat het gebruik van orale anticoagulantia of NSAID’s, door stimulatie van bloedingen uit premaligne laesies, een voordelig effect heeft door meer ‘true (i)FOBT positives’ te veroorzaken in gebruikers van deze medicijnen. Bewijskracht literatuur: study quality level 1 (SORT).
Endoscopie/colonoscopie en het risico op bloedingen
De gevonden literatuur betreft NIET in het bijzonder patiënten met hemofilie maar geeft richting aan het inschatten van risico’s van colonoscopie. Het risico op bloedingen verschilt tussen de diagnostische en therapeutische endoscopische procedures. De ‘American Society for Gastrointestinal Endoscopy’ (Acosta, 2016) en de ‘European Society of Gastrointestinal Endoscopy’ (Veitch, 2016) hebben richtlijnen gepubliceerd voor patiënten met antitrombotische medicijnen en maken een onderscheid tussen laag risico en hoog risico procedures. Diagnostische colonoscopie met of zonder het nemen van biopten valt onder de laag risico procedures wat betreft bloedingen. Colonoscopie met poliepectomie valt onder de hoog risico procedures. Het risico op bloedingen na poliepectomie in de algemene bevolking varieert van 0,3-10% afhankelijk van o.a. de grootte van de poliep, locatie, morfologie en de resectie techniek (Acosta, 2016). In het artikel van de ‘European Society of Gastrointestinal Endoscopy’ (Veitch, 2016) worden richtlijnen voor het management van verschillende antitrombotische behandelingen bij laag en hoog risico endoscopische procedures weergegeven. Heel algemeen gezegd kan bij laag risico procedures de antitrombotische therapie voortgezet worden en wordt bij hoog risico procedures antitrombotische therapie tijdelijk gestopt (afhankelijk van het risico op trombotische complicaties).
De ‘American Society for Gastrointestinal Endoscopy’ (Fisher, 2011) beschrijft in een artikel uit 2011 de complicaties van colonoscopie in de algemene populatie: serious adverse event rate was 2,8 per 1000 procedures. Meer dan 85% van de ernstige complicaties treden op bij colonoscopie met poliepectomie. Poliepectomie is geassocieerd met een 7-voudig verhoogd risico op perforatie of bloeding. De afmeting van de verwijderde poliep, het aantal verwijderde poliepen, histologie en gebruik van anticoagulantia zijn risicofactoren voor bloeding (Fischer, 2011). Bewijskracht literatuur: study quality level 2 (SORT).
Colonoscopie bij patiënten met hemofilie
De gevonden artikelen over patiënten met hemofilie die colonoscopie ondergaan beschrijven enkele case-series (Tintillier, 2013; Davis, 2013; Fogarty, 2010; Tomaszewski, 2019) en “expert-opinie” (Astermark, 2012; Fogarty, 2010; van Os, 1999). Alle ‘experts’ zijn van mening dat colonoscopieën uitgevoerd kunnen worden bij hemofilie patiënten indien de stollingsfactordeficiëntie volledig gecorrigeerd wordt. De optimale behandeling (dosis en duur) van de stollingsfactordeficiëntie is afhankelijk van de ernst van de hemofilie en van het type endoscopische interventie. Pre-procedureel streefwaarde stollingsfactor VIII/IX activiteit 80-120 IE/dLl, en post-procedureel suppleren 1-3 dagen tot 4-8 dagen na een poliepectomie (Tintillier, 2013; Astermark, 2012). Tranexaminezuur kan worden toegediend bij invasieve procedures (Astermark, 2012). Bewijskracht literatuur: study quality level 3 (SORT)
Referenties
Bewijskracht literatuur
Level 1-3, per paragraaf toegelicht in de samenvatting van de literatuur (resultaten) .
Zoekverantwoording
De verschillende onderdelen van de zoekstrategie (zie Tabel 11.3.1) zijn in verschillende combinaties als zoekopdrachten in PubMed gebruikt.
Tabel 11.3.1. Opbouw zoekstrategie PubMed
|
Keyword |
MeSH database |
Title and Abstract (tiab) |
Combineren (MesH/tiab) |
|
Hemophilia |
(“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh] |
(hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract] |
(((“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh])) OR ((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]) |
|
Bleeding disorder |
“Blood Coagulation Disorders”[Mesh] |
(blood coagulation disorder*[Title/Abstract]) OR bleeding disorder*[Title/Abstract] |
((“Blood Coagulation Disorders”[Mesh]) OR blood coagulation disorder*[Title/Abstract]) OR bleeding disorder*[Title/Abstract] |
|
Fecal occult blood test |
Geen Mesh term
|
(((((((fecal occult blood[Title/Abstract]) OR FOB[Title/Abstract]) OR fecal occult blood test[Title/Abstract]) OR FOBT[Title/Abstract]) OR immunological fecal occult blood test[Title/Abstract]) OR (i)FOBT[Title/Abstract]) OR fecal immunological test[Title/Abstract]) OR FIT[Title/Abstract] |
|
|
Colon carcinoma |
“Colorectal Neoplasms”[Mesh] |
(((((colon cancer[Title/Abstract]) OR colon carcinoma[Title/Abstract]) OR colorectal cancer[Title/Abstract]) OR colorectal carcinoma[Title/Abstract]) OR cancer[Title/Abstract]) OR malignan*[Title/Abstract] |
(“Colorectal Neoplasms”[Mesh]) OR ((((((colon cancer[Title/Abstract]) OR colon carcinoma[Title/Abstract]) OR colorectal cancer[Title/Abstract]) OR colorectal carcinoma[Title/Abstract]) OR cancer[Title/Abstract]) OR malignan*[Title/Abstract]) |
|
Gastrointestinal bleeding |
“Gastrointestinal Hemorrhage”[Mesh] |
(((((gastrointestinal hemorrhage[Title/Abstract]) OR gastrointestinal bleeding[Title/Abstract]) OR GI bleeding[Title/Abstract]) OR colorectal bleeding[Title/Abstract]) OR rectal bleeding[Title/Abstract] |
((“Gastrointestinal Hemorrhage”[Mesh]) OR (((((gastrointestinal hemorrhage[Title/Abstract]) OR gastrointestinal bleeding[Title/Abstract]) OR GI bleeding[Title/Abstract]) OR colorectal bleeding[Title/Abstract]) OR rectal bleeding[Title/Abstract])) |
|
Antithrombotic |
(“Anticoagulants”[Mesh]) OR “Platelet Aggregation Inhibitors”[Mesh] |
((antithrombotic*[Title/Abstract]) OR anticoagulant*[Title/Abstract]) OR antiplatelet |
(((“Anticoagulants”[Mesh]) OR “Platelet Aggregation Inhibitors”[Mesh])) OR (((antithrombotic*[Title/Abstract]) OR anticoagulant*[Title/Abstract]) OR antiplatelet) |
|
Meta-analysis |
“Meta-Analysis” [Publication Type] |
meta-analysis[Title/Abstract] |
(“Meta-Analysis” [Publication Type]) OR meta-analysis[Title/Abstract] |
|
Colonoscopy |
“Endoscopy, Gastrointestinal”[Mesh]: (colonoscopy valt onder deze Mesh term) |
(((coloscopy[Title/Abstract]) OR colonoscopy[Title/Abstract]) OR colonscopy[Title/Abstract]) OR endoscop*[Title/Abstract] |
(“Endoscopy, Gastrointestinal”[Mesh]) OR ((((coloscopy[Title/Abstract]) OR colonoscopy[Title/Abstract]) OR colonscopy[Title/Abstract]) OR endoscop*[Title/Abstract]) |
Zoekopdracht (i)FOBT bij patiënten met hemofilie
(((((“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh])) OR ((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]))) AND ((((((((fecal occult blood[Title/Abstract]) OR FOB[Title/Abstract]) OR fecal occult blood test[Title/Abstract]) OR FOBT[Title/Abstract]) OR immunological fecal occult blood test[Title/Abstract]) OR (i)FOBT[Title/Abstract]) OR fecal immunological test[Title/Abstract]) OR FIT[Title/Abstract]).
Deze zoekopdracht resulteert in 49 publicaties. De resultaten van de zoekstrategie zijn gescreend op titel en samenvatting. Na deze eerste screening blijven er geen publicaties over welke mogelijk interessant zijn voor onze zoekvragen.
Om de mogelijkheid te onderzoeken dat bij andere stollingsstoornissen wel iets bekend is over FOBT, is een nieuwe zoekopdracht gemaakt met het bredere ‘keyword’ ‘bleeding disorder’.
((((“Blood Coagulation Disorders”[Mesh]) OR blood coagulation disorder*[Title/Abstract]) OR bleeding disorder*[Title/Abstract])) AND ((((((((fecal occult blood[Title/Abstract]) OR FOB[Title/Abstract]) OR fecal occult blood test[Title/Abstract]) OR FOBT[Title/Abstract]) OR immunological fecal occult blood test[Title/Abstract]) OR (i)FOBT[Title/Abstract]) OR fecal immunological test[Title/Abstract]) OR FIT[Title/Abstract]).
Deze zoekopdracht levert 100 publicaties op, waarna na screening van titel en samenvatting geen publicaties overblijven.
Zoekopdracht hemofilie en maligniteiten
(((((“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh])) OR ((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]))) AND ((“Colorectal Neoplasms”[Mesh]) OR ((((((colon cancer[Title/Abstract]) OR colon carcinoma[Title/Abstract]) OR colorectal cancer[Title/Abstract]) OR colorectal carcinoma[Title/Abstract]) OR cancer[Title/Abstract]) OR malignan*[Title/Abstract]))
De 589 artikelen met brede zoekopdracht zijn niet allemaal gescreend, echter een aantal relevante en geciteerde artikelen zijn interessant bevonden voor de zoekvraag “coloncarcinoom bij patiënten met hemofilie”.
Zoekopdracht hemofilie en gastrointestinale bloedingen
(((((“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh])) OR ((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]))) AND ((“Gastrointestinal Hemorrhage”[Mesh]) OR (((((gastrointestinal hemorrhage[Title/Abstract]) OR gastrointestinal bleeding[Title/Abstract]) OR GI bleeding[Title/Abstract]) OR colorectal bleeding[Title/Abstract]) OR rectal bleeding[Title/Abstract]))
Totaal 268 artikelen. Na exclusie van case reports en screening op titel en samenvatting blijven er 12 artikelen over.
Zoekopdrachten met betrekking tot de invloed van antistolling op de (i)FOBT
((((((((((fecal occult blood[Title/Abstract]) OR FOB[Title/Abstract]) OR fecal occult blood test[Title/Abstract]) OR FOBT[Title/Abstract]) OR immunological fecal occult blood test[Title/Abstract]) OR (i)FOBT[Title/Abstract]) OR fecal immunological test[Title/Abstract]) OR FIT[Title/Abstract])) AND ((((“Anticoagulants”[Mesh]) OR “Platelet Aggregation Inhibitors”[Mesh])) OR (((antithrombotic*[Title/Abstract]) OR anticoagulant*[Title/Abstract]) OR antiplatelet))) AND ((“Meta-Analysis” [Publication Type]) OR meta-analysis[Title/Abstract]).
In deze eerste zoekopdracht naar de invloed van antistolling op de (i)FOBT is gezocht naar meta-analyses, om een eerste beeld te verkrijgen over de beschikbare literatuur. De zoekopdracht resulteert in 12 publicaties, welke gescreend zijn op titel en samenvatting. Drie artikelen zijn mogelijk interessant voor onze zoekvraag: Gandhi S, 2013; Nieuwenburg, 2018; de Klerk, 2018.
Zoekopdracht colonoscopie bij hemofilie patiënten
(((((“Hemophilia A”[Mesh]) OR “Hemophilia B”[Mesh])) OR ((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]))) AND ((“Endoscopy, Gastrointestinal”[Mesh]) OR ((((coloscopy[Title/Abstract]) OR colonoscopy[Title/Abstract]) OR colonscopy[Title/Abstract]) OR endoscop*[Title/Abstract])).
Deze zoekopdracht resulteert in 74 publicaties. De resultaten van de zoekstrategie zijn gescreend op titel en samenvatting. Na deze screening blijven er 6 publicaties over welke mogelijk interessant zijn voor onze zoekvraag. Na het controleren van de referenties blijken er 2 publicaties mogelijk van belang die niet gevonden zijn met de initiële zoekstrategie in PubMed: Van Os, 1999; en Astermark, 2012.
Evidence tabellen
N.v.t.
Overwegingen
De originele zoekvraag over de waarde van de (immunologisch) fecaal occult bloed test in patiënten met hemofilie levert slechts een artikel op met een bruikbare “expert opinie”.
Met betrekking tot de deelvragen die behulpzaam zijn bij beantwoorden van de hoofdvraag:
Colonoscopie in het kader van het bevolkingsonderzoek darmkanker bij hemofilie patiënten
In het ‘NHG-standpunt Bevolkingsonderzoek darmkanker’ (Wiersma, 2013) wordt niets beschreven over patiënten met een stollingsstoornis. Wel wordt beschreven wat er gebeurt nadat de uitslag van de (i)FOBT positief blijkt: ‘De huisarts komt in beeld als de uitslag van de (i)FOBT positief blijkt. Met de screeningsorganisaties is afgesproken dat de huisarts van de betrokken patiënt 1-2 werkdagen eerder dan de patiënt bericht krijgt, zodat hij of zij de gelegenheid heeft de patiënt van de uitslag op de hoogte te brengen en de betekenis daarvan kan toelichten en verduidelijken. Met het bericht van een positieve uitslag en de afspraak voor een intakegesprek krijgt de patiënt tevens het verzoek contact op te nemen met zijn huisarts, zodat laatstgenoemde relevante medische informatie kan toespelen aan het colonoscopie-centrum (als de huisarts al niet zelf contact had opgenomen met de patiënt). Bij relevante medische informatie gaat het vooral om belangrijke comorbiditeit, zoals cardiovasculaire problemen, eerdere ingrepen in de buik en gebruik van antistolling waarmee rekening gehouden moet worden bij de colonoscopie en eventuele latere operatie.’
Coloncarcinoom bij patiënten met hemofilie
Er lijken geen aanwijzingen te zijn dat colorectaal carcinoom een hogere of lagere incidentie heeft bij patiënten met hemofilie vergeleken met de algemene bevolking. Er is om deze reden niet op voorhand indicatie voor een andere wijze van screening op deze vorm van maligniteit dan in de algemene bevolking.
Gastro-intestinale bloedingen in patiënten met hemofilie
Er zijn vooral aanwijzingen dat bloedingen uit de bovenste tractus digestivus vaker voorkomen bij patiënten met hemofilie. Over bloedingen uit de onderste tractus digestivus zijn wij niet goed geïnformeerd. Er is om deze reden niet op voorhand indicatie voor een andere wijze van screening op colorectaal carcinoom dan in de algemene bevolking.
Wat is de invloed van antistolling op de (i)FOBT
Er zou een hogere ‘positivity rate’ in patiënten met orale anticoagulantia of NSAID-gebruik zijn. Ook voor patiënten met hemofilie zou deze hogere ‘positivity rate’ kunnen gelden. Het gevolg is dat hemofilie patiënten in het bevolkingsonderzoek darmkanker vaker een colonoscopie zouden ondergaan dan personen uit de algemene populatie, met het gebruik van stollingsfactoren en het risico op complicaties van dien. Voorts kan worden geconcludeerd dat de nauwkeurigheid van de (i)FOBT bij het opsporen van ‘advanced neoplasia’ en colorectaal carcinoom vergelijkbaar is tussen patiënten zonder en met orale anticoagulantia of NSAID’s. Daarnaast blijkt de positief voorspellende waarde van de (i)FOBT in personen met en zonder antitrombotische medicijnen vergelijkbaar (en voor DAPT-gebruikers zelfs hoger). Dit zou kunnen betekenen dat het gebruik van orale anticoagulantia of NSAID’s, door stimulatie van bloedingen uit premaligne laesies, een voordelig effect heeft door meer ‘true (i)FOBT positives’ te veroorzaken in gebruikers van deze medicijnen. Ook deze hypothese zou bij patiënten met hemofilie kunnen gelden, waardoor men zou kunnen overwegen het verhoogde aantal colonoscopieën verantwoord te vinden.
Endoscopie/colonoscopie en het risico op bloedingen
De gevonden literatuur betreft NIET in het bijzonder patiënten met hemofilie maar geeft richting aan het inschatten van risico’s van colonoscopie. Diagnostische colonoscopie met of zonder het nemen van biopten valt onder de laag risico procedures wat betreft bloedingen. Colonoscopie met poliepectomie valt onder de hoog risico procedures. Het risico op bloedingen is afhankelijk van o.a. de grootte van de poliep, locatie, morfologie en de resectie techniek.
Colonoscopie in het kader van het bevolkingsonderzoek darmkanker bij hemofilie patiënten
Hemofilie is “relevante comorbiditeit” in het kader van het ‘NHG-standpunt Bevolkingsonderzoek darmkanker’ en leidt tot de overweging dat de hemofilie patiënt een colonoscopie zou moeten ondergaan in het ziekenhuis waar het hemofiliebehandelcentrum van de patiënt gevestigd is.
Wat zijn contra-indicaties voor het gebruik van DDAVP bij de oudere patiënt met hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Desmopressine (1-deamino-8-D-arginine-vasopressine, DDAVP) veroorzaakt een stijging in de stollingsfactor VIII (FVIII) en Von Willebrand Factor (VWF) plasmaconcentraties bij patiënten met milde hemofilie A of de ziekte van Von Willebrand (Mannucci, 1997; Franchini, 2007; Leissinger, 2014). Van de hemostatische effecten van DDAVP wordt ook gebruik gemaakt bij draagsters van hemofilie A met een verlaagd stollingsfactor VIII gehalte en bij patiënten met een trombocytopathie (Franchini, 2007; Leissinger, 2014). DDAVP wordt gebruikt ter behandeling van lichte bloedingen of ter preventie van bloedingen bij kleine ingrepen (Farmacotherapeutisch Kompas; Mannucci, 1997).
DDAVP is een synthetische afgeleide van vasopressine (Farmacotherapeutisch Kompas) en is oorspronkelijk ontwikkeld voor de behandeling van diabetes insipidus (Mannucci, 1997). Vasopressine heeft een antidiuretisch effect via V2-receptoren in de nieren en wordt daarom ook wel antidiuretisch hormoon (ADH) genoemd. Activering van V2-receptoren leidt ook tot een verhoging van intracellulaire concentraties van cyclisch adenosine-monofosfaat, met als gevolg exocytose van VWF uit Weibel-Palade lichaampjes van endotheelcellen in de circulatie (Mannucci 1997; Özgӧnenel, 2006; Franchini, 2010). Daarnaast veroorzaakt vasopressine contractie van glad spierweefsel (via V1-receptoren), met als gevolg een lichte stijging van de bloeddruk en constrictie van de darmen en coronair arteriën (Farmacotherapeutische Kompas). DDAVP activeert voornamelijk V2-receptoren en heeft een sterkere en langer aanhoudende antidiuretische werking dan vasopressine (Farmacotherapeutisch Kompas; Özgӧnenel, 2006).
Binding van DDAVP aan V2-receptoren leidt tot een toename van de tubulaire resorptie van water. Belangrijke mogelijke gevolgen, die niet frequent vóórkomen, zijn hyponatriëmie en uiteindelijk waterintoxicatie, waarbij in ernstige gevallen convulsies en bewusteloosheid optreden. Veelvoorkomende milde bijwerkingen door binding aan V2-receptoren, welke vaak het gevolg zijn van snelle infusie, zijn: blozen, tachycardie en hoofdpijn (Farmacotherapeutisch Kompas; Mannucci, 1997; Leissinger, 2014; Stoof, 2016). Omdat V1-receptoren minimaal worden geactiveerd door DDAVP, komen bijwerkingen door gastro-intestinale contracties of hypertensie weinig voor (Mannucci, 1997; Leissinger, 2014).
Conclusies
|
SORT Grade |
Conclusie |
|
N.v.t. |
Er is geen literatuur gevonden over een leeftijdsgrens waarboven het gebruik van DDAVP gecontra-indiceerd is. |
|
C |
DDAVP kan leiden tot waterretentie en hyponatriëmie. Dit geldt vooral bij ouderen >65 jaar en bij patiënten met cardiale insufficiëntie. (Mannucci, 1997; Farmacotherapeutisch Kompas) |
|
C |
Er is een verband tussen DDAVP toediening en het optreden van ischemische vasculaire events. (Castaman, 2008) |
|
C |
Het effect van DDAVP op de bloeddruk is eerder verlagend dan verhogend. (Castaman, 2008; Leissinger, 2014; Stoof, 2016) |
Samenvatting literatuur
Resultaten
In de ‘Guidelines for the management of hemophilia’ van de World Federation of Hemophilia (WFH) uit 2012 (Srivastava in Hemophilia WFH guidelines, 2012) wordt aanbevolen om de plasma-osmolaliteit of natriumconcentratie te meten wanneer er herhaaldelijk DDAVP wordt toegediend (Mannucci, 1997; Sica, 2006). Hierbij wordt niet specifiek een leeftijdsgroep genoemd. Ook in de Nederlandse richtlijn uit 2009 wordt aanbevolen het serum natrium gehalte te controleren wanneer DDAVP gedurende meerdere dagen wordt gegeven (NVHB richtlijn, 2009). Overmatige vochtinname (per os of intraveneus) moet worden vermeden de eerste 8-12 uur na toediening, vanwege het risico op waterretentie en hyponatriëmie (NVHB richtlijn, 2009). Hiermee moet ook rekening gehouden worden tijdens de peri- en postoperatieve situatie (NVHB richtlijn, 2009).
Het Farmacotherapeutisch Kompas vermeldt dat er meer kans is op hyponatriëmie bij ouderen vanaf 65 jaar en dat bij langdurige behandeling het serum natrium gehalte regelmatig gecontroleerd dient te worden. Bij tekenen van waterretentie/hyponatriëmie (hoofdpijn, misselijkheid, braken, maagpijn, spierkrampen, duizeligheid, verwardheid, gewichtstoename, malaise, convulsies en coma) moet de behandeling met DDAVP meteen (tijdelijk) gestaakt worden tot volledig herstel is bereikt. Bij hervatting van de behandeling, moet de beperking van de vloeistofinname strikt aangehouden worden en het serum natrium gehalte worden gecontroleerd (Farmacotherapeutisch Kompas).
Prostaglandinesynthetaseremmers en oxytocine versterken het antidiuretisch effect van DDAVP door inductie van waterretentie/hyponatriëmie. Bij gelijktijdig gebruik van middelen die de afgifte van ADH verstoren, zoals tricyclische antidepressiva, SSRI’s, chloorpromazine, carbamazepine en mogelijk sulfonylureumderivaten neemt de kans op waterintoxicatie toe. Bij gelijktijdig gebruik van loperamide neemt de kans op waterretentie/hyponatriëmie toe, door een verdrievoudiging van de plasmaconcentratie van DDAVP. Gelijktijdig gebruik met dimeticon kan de resorptie verminderen. Lithium kan het antidiuretisch effect verminderen. Een lithium geïnduceerde nefrogene diabetes insipidus kan worden gemaskeerd tijdens gebruik van DDAVP (Farmacotherapeutisch Kompas). De oudere patiënt, en naar verwachting ook de oudere hemofilie patiënt, gebruikt vaak meerdere medicijnen. Van belang is de mogelijkheid op interacties na te gaan wanneer een oudere patiënt met hemofilie behandeld dient te worden met DDAVP.
Een aantal gevallen van arteriële trombose, waaronder myocard infarct, na toediening van DDAVP is vermeld in de literatuur (Bond, 1988; Byrnes, 1988; Manucci, 1989; van Dantzig, 1989; Girolami, 2006). Voorzichtigheid is geboden bij het gebruik van DDAVP in hemofilie patiënten met een voorgeschiedenis van, of een hoog risico op, hart- en vaatziekten (Farmacotherapeutisch Kompas; WFH guidelines, 2012; Castaman, 2008).
Voor behandeling met DDAVP dient het effect van de DDAVP toediening op de verhoging van het stollingsfactor VIII gehalte en het ontstaan van bijwerkingen gecontroleerd te worden (WFH guidelines, 2012; Mannucci, 1997; Castaman, 2009). De DDAVP respons is zeer verschillend tussen individuen, maar binnen dezelfde persoon is het effect van DDAVP consistent (Castaman, 2009; Rodeghiero, 1989). In een studie van Castaman et al. (Castaman, 2009) zijn moleculaire en fenotypische determinanten van de DDAVP respons in volwassenen met milde hemofilie onderzocht. Vijftig patiënten met een mediane leeftijd van 40 jaar (range 19-69) zijn geïncludeerd in de studie. In het volledige cohort, bleken post-DDAVP FVIII:C piek waarden positief geassocieerd met FVIII:C basale waarden (p<0.0001), maar niet met Von Willebrand Factor antigen (VWF:Ag) basale of piek waarden, leeftijd of bloedgroep (multivariate analyse). Echter, in een subgroep analyse van de patiënten met een geïdentificeerde mutatie (n=41) bleek leeftijd wel positief geassocieerd (p=0.03) met FVIII:C piek waarden. Ook de FVIII:C halfwaardetijd was positief geassocieerd met leeftijd (p=0.004). De associatie tussen factor VIII:C piekwaarde na DDAVP infusie en FVIII:C basale waarden alsmede stijging van piekwaarden met toename van de leeftijd werd beschreven door Mauser et al. (Mauser-Bunschoten, 2013).
Als contra-indicaties (onafhankelijk van leeftijd) voor het gebruik van DDAVP worden in het Farmacotherapeutisch Kompas vermeld:
Referenties
Bewijskracht literatuur
Level 3, consistent.
Zoekverantwoording
Hieronder volgt het systematische literatuuronderzoek naar DDAVP bij de oudere hemofilie patiënt.
Pagina-einde
Table 11.4.1. Opbouw zoekstrategie PubMed 25-01-2019
|
Keyword |
MeSH database |
Title and Abstract (tiab) |
Combineren (MesH/tiab) |
|
Hemophilia |
((“Hemophilia A”[Mesh]) OR “von Willebrand Diseases”[Mesh]) OR “Blood Coagulation Disorders”[Mesh] |
(((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]) OR von Willebrand disease[Title/Abstract]) OR congenital bleeding disorder*[Title/Abstract] |
((((“Hemophilia A”[Mesh]) OR “von Willebrand Diseases”[Mesh]) OR “Blood Coagulation Disorders”[Mesh])) OR ((((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]) OR von Willebrand disease[Title/Abstract]) OR congenital bleeding disorder*[Title/Abstract]) |
|
Desmopressin |
“Deamino Arginine Vasopressin”[Mesh] |
((desmopressin[Title/Abstract]) OR DDAVP[Title/Abstract]) OR 1-deamino-8-D-arginine-vasopressin[Title/Abstract] |
(“Deamino Arginine Vasopressin”[Mesh]) OR (((desmopressin[Title/Abstract]) OR DDAVP[Title/Abstract]) OR 1-deamino-8-D-arginine-vasopressin[Title/Abstract]) |
|
Age |
(“Aging”[Mesh]) OR “Aged”[Mesh] |
(((aging[Title/Abstract]) OR ageing[Title/Abstract]) OR elderly[Title/Abstract]) OR old*[Title/Abstract] |
(((“Aging”[Mesh]) OR “Aged”[Mesh])) OR ((((aging[Title/Abstract]) OR ageing[Title/Abstract]) OR elderly[Title/Abstract]) OR old*[Title/Abstract]) |
|
Safety |
(“Safety”[Mesh]) OR “Drug-Related Side Effects and Adverse Reactions”[Mesh] |
((((safety[Title/Abstract]) OR adverse event*[Title/Abstract])) OR side effect*[Title/Abstract]) |
(((“Safety”[Mesh]) OR “Drug-Related Side Effects and Adverse Reactions”[Mesh])) OR (((((safety[Title/Abstract]) OR adverse event*[Title/Abstract])) OR side effect*[Title/Abstract])) |
De onderdelen van de zoekstrategie (Tabel 11.4.1) zijn op verschillende manieren gecombineerd.
Zoekopdracht 1:
De zoekstrategiën van alle 4 de ‘keywords’ zijn gecombineerd:
((((((((“Hemophilia A”[Mesh]) OR “von Willebrand Diseases”[Mesh]) OR “Blood Coagulation Disorders”[Mesh])) OR ((((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]) OR von Willebrand disease[Title/Abstract]) OR congenital bleeding disorder[Title/Abstract]))) AND ((“Deamino Arginine Vasopressin”[Mesh]) OR (((desmopressin[Title/Abstract]) OR DDAVP[Title/Abstract]) OR 1-deamino-8-D-arginine-vasopressin[Title/Abstract]))) AND ((((“Aging”[Mesh]) OR “Aged”[Mesh])) OR ((((aging[Title/Abstract]) OR ageing[Title/Abstract]) OR elderly[Title/Abstract]) OR old*[Title/Abstract]))) AND ((((“Safety”[Mesh]) OR “Drug-Related Side Effects and Adverse Reactions”[Mesh])) OR (((safety[Title/Abstract]) OR adverse event*[Title/Abstract]) OR side effect*[Title/Abstract]))
Deze zoekopdracht resulteert in 28 publicaties. De resultaten van de zoekstrategie zijn gescreend op titel en samenvatting. Na deze eerste screening blijven er 3 publicaties over welke mogelijk interessant zijn voor onze zoekvragen (Tabel 11.4.2).
Zoekopdracht 2:
De zoekstrategiën van de volgende ‘keywords’ zijn gecombineerd: ‘hemophilia’, ‘demopressin’ en ‘safety’.
(((((((“Hemophilia A”[Mesh]) OR “von Willebrand Diseases”[Mesh]) OR “Blood Coagulation Disorders”[Mesh])) OR ((((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]) OR von Willebrand disease[Title/Abstract]) OR congenital bleeding disorder*[Title/Abstract]))) AND ((“Deamino Arginine Vasopressin”[Mesh]) OR (((desmopressin[Title/Abstract]) OR DDAVP[Title/Abstract]) OR 1-deamino-8-D-arginine-vasopressin[Title/Abstract]))) AND ((((“Safety”[Mesh]) OR “Drug-Related Side Effects and Adverse Reactions”[Mesh])) OR (((((safety[Title/Abstract]) OR adverse event*[Title/Abstract])) OR side effect*[Title/Abstract])))
Deze zoekopdracht resulteert in 134 publicaties. De resultaten van de zoekstrategie zijn gescreend op titel en samenvatting. Na deze eerste screening blijven er 8 publicaties, waaronder de 3 publicaties gevonden met zoekopdracht 1 (Tabel 11.4.2), over welke mogelijk interessant zijn voor onze zoekvragen (Tabel 11.4.3).
Zoekopdracht 3:
De zoekstrategiën van de volgende ‘keywords’ zijn gecombineerd: ‘hemophilia’, ‘demopressin’ en ‘age’.
(((((((“Hemophilia A”[Mesh]) OR “von Willebrand Diseases”[Mesh]) OR “Blood Coagulation Disorders”[Mesh])) OR ((((hemophilia*[Title/Abstract]) OR haemophilia*[Title/Abstract]) OR von Willebrand disease[Title/Abstract]) OR congenital bleeding disorder[Title/Abstract]))) AND ((“Deamino Arginine Vasopressin”[Mesh]) OR (((desmopressin[Title/Abstract]) OR DDAVP[Title/Abstract]) OR 1-deamino-8-D-arginine-vasopressin[Title/Abstract]))) AND ((((“Aging”[Mesh]) OR “Aged”[Mesh])) OR ((((aging[Title/Abstract]) OR ageing[Title/Abstract]) OR elderly[Title/Abstract]) OR old*[Title/Abstract]))
Deze zoekopdracht resulteert in 249 publicaties. De resultaten van de zoekstrategie zijn gescreend op titel en samenvatting. Er zijn geen andere mogelijk interessante publicaties gevonden dat degenen die al gevonden waren met zoekopdracht 1 en 2.
Evidence tabellen
Tabel 11.4.2. Resultaten zoekopdracht 1
Tabel 11.4.3. Resultaten zoekopdracht 2
Overwegingen
Tot welke leeftijd kunnen patiënten met hemofilie veilig behandeld worden met DDAVP?
Met het gevonden bewijs kan er niet gesteld worden dat hemofilie patiënten vanaf een bepaalde leeftijd niet meer behandeld zouden mogen worden met DDAVP. De oudere patiënt heeft meer risico op het ontwikkelen van hyponatriëmie bij de behandeling met DDAVP, met name als er meer dan één dosis gegeven wordt. Het Farmacotherapeutisch Kompas stelt dat dit geldt voor patiënten vanaf 65 jaar. Hier wordt echter geen referentie voor gegeven. Wanneer gedurende meer dagen DDAVP wordt gegeven, dient het serum natrium gehalte gecontroleerd te worden. Overmatige vochtinname moet vermeden worden. Bij tekenen van waterretentie/hyponatriëmie moet de behandeling gestopt worden. De review van Leissinger (Leissinger, 2014) geeft aan dat hyponatriëmie-gerelateerde bijwerkingen worden veroorzaakt door het antidiuretisch effect van DDAVP, welke 10x hoger is met intraveneus dan met intranasaal gebruik. Er wordt in deze review geen verdere onderbouwing of bewijs hiervoor gegeven.
Wat zijn de contra-indicaties voor het gebruik van DDAVP bij de ouder wordende patiënt met hemofilie?
Absolute contra-indicaties specifiek voor de ouder wordende hemofilie patiënt worden niet vermeld in de literatuur. Voorzichtigheid is geboden bij het gebruik van DDAVP in hemofilie patiënten met een voorgeschiedenis van, of een hoog risico op, hart- en vaatziekten (Farmacotherapeutisch Kompas; WFH guidelines, 2012; Castaman, 2008). De oudere patiënt, en naar verwachting ook de oudere hemofilie patiënt, gebruikt vaak meer medicijnen/polyfarmacie. Van belang is de mogelijkheid op interacties na te gaan wanneer overwogen wordt een oudere patiënt met hemofilie te behandelen met DDAVP.
Het besluit om anticoagulantia en trombocytenaggregatieremmers therapie te starten bij patiënten met hemofilie berust, net als in de algemene populatie, op een afweging tussen baten (bescherming tegen trombotische events) en schade (bloedingsrisico). Bij hemofilie is altijd, in meer of mindere mate, het bloedingsrisico hoger dan in de algemene populatie. Of ook de winst door bescherming tegen trombotisch events anders is, hangt af van de situatie: patiënten met hemofilie hebben soms wel, maar soms ook niet een lager risico op trombotische events.
In deze richtlijn wordt de algemene uitgangsvraag: “Wat is het beste antitrombotisch beleid bij patiënten met hemofilie?” uitgewerkt in een aantal subvragen die klinisch relevant zijn. De richtlijn geeft geen adviezen voor patiënten die een remmer hebben. De adviezen voor patiënten met hemofilie gelden ook voor draagsters van hemofilie.
In deze uitgangsvraag wordt de volgende terminologie aangehouden:
Onder welke omstandigheden kunnen antitrombotica veilig worden gegeven bij de behandeling van acuut coronair syndroom in patiënten met hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Deze vraag betreft een situaties waar trombose al is opgetreden De vraag naar het risico op acuut coronair syndroom bij hemofilie is dus minder relevant. Voor deze vraagstelling wordt alleen naar het bloedingsrisico gekeken.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
Behandeling van een acuut coronair syndroom met percutane coronair interventie volgens de actuele cardiologie richtlijnen kan in patiënten met hemofilie veilig plaatsvinden onder adequate substitutie therapie. (Kulkarni, 2005) |
|
N.v.t. |
Het stollingsfactor gehalte waarbij therapeutische antistolling veilig kan plaatsvinden is in de literatuur niet uitgezocht. |
Samenvatting literatuur
De prevalentie van ischemische hartziekten (angina pectoris, acuut coronair syndroom) in hemofilie patiënten ouder dan 60 jaar is 15% (Kulkarni, 2005). De cumulatieve incidentie van niet fataal myocardinfarct is 1,7% in ernstige hemofilie patiënten, 3,6% in niet ernstige patiënten en 4% in algemene populatie (Fransen van de Putte, 2012). De behandeling van acuut coronair syndroom (ACS) omvat onder andere het verrichten van een percutane coronair interventie (PCI), waarbij patiënten tijdens de procedure hoge doseringen anticoagulantia en trombocyten aggregatieremmers krijgen. Deze behandeling is in hemofilie patiënten niet gerandomiseerd uitgezocht. Een artikel beschrijft de toepasbaarheid van de Europese cardiologie richtlijn voor ACS op de populatie van hemofilie patiënten(Staritz, 2013), daarnaast is er een lokale Nederlandse richtlijn prospectief geëvalueerd (Schutgens, 2009; Tuinenburg, 2013). Er zijn meerdere literatuur reviews en case reports gepubliceerd. De meest recente systematische literatuur review is uit 2016, hierin werd onder andere ook de prospectieve evaluatie van de lokale Nederlandse richtlijn meegenomen. De auteurs beschreven in totaal 54 patiënten (20 patiënten ernstig hemofilie A, 5 met matige-ernstige hemofilie A en 19 met milde hemofilie A, 1 patiënt onbekend) met acuut coronair syndroom waarvan 38 patiënten een PCI ondergingen. Vierentwintig van deze patiënten kregen antitrombotica in de acute situatie met een bolus ongefractioneerde heparine gecombineerd met hoge dosis trombocyten aggregatieremmers , twee patiënten kregen low molecular weight heparin (LMWH), vier patiënten bivalirudin en acht patiënten kregen geen antitrombotica. In 33 patiënten was er sprake van adequate factor substitutie (> 70IE/dL of > 80 IE/dL). Bij in totaal drie patiënten (6%) trad een ernstige bloeding op. Twee patiënten kregen een bloeding ter plaatse van de punctieplek van de arterie femoralis. Een patiënt ontwikkelde een trauma gerelateerde intramusculaire bloeding (Boehnel, 2017).
Referenties
Bewijskracht literatuur
Level 3
Zoeken en selecteren
20-09-2018 Pubmed: (((“acute coronary syndrome”[Mesh] OR “Percutaneous coronary intervention”[MESH]))) AND ((“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab])). Aantal hits: 33
20-09-2018 Pubmed: (“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab]) AND (“Anticoagulation” [Pharmacological Action] OR “Anticoagulation” [Mesh] OR “Anticoagulants”[Mesh] OR anticoagu*[tiab] OR antico*[tiab] OR anti-coagu*[tiab] OR anticoagula*[tiab] OR heparin[tiab] OR low molecular weight*[tiab] OR oral anticoagul*[tiab] OR LMWH*[tiab] OR bivalirudin*[tiab] OR VKA*[tiab] OR vitamin K antagonists*[tiab])
AND (“Hemorrhage”[Mesh] OR hemorrhag*[tiab] OR haemorrhag*[tiab] OR bleeding[tiab]). Aantal hits: 478.
Vanuit de Pubmed strategie en vanuit de referentielijsten van de geselecteerde artikelen of vanuit de richtlijnen werden in totaal 6 full text artikelen geselecteerd voor deze uitgangsvraag.
Daarnaast werd de Nederlandse richtlijn Antitrombotisch beleid van de Nederlandse Internisten Vereniging geraadpleegd.
Overwegingen
De literatuur laat zien dat behandeling van een acuut coronair syndroom met percutane coronair interventie volgens de actuele cardiologie richtlijnen in patiënten met hemofilie veilig kan plaatsvinden onder adequate substitutie therapie. Het is hierbij van belang dat interventies en het voorschrijven van antitrombotica bij hemofilie patiënten met een acuut coronair syndroom gebeuren in nauwe samenspraak tussen de cardioloog en hemofiliebehandelaar. Er worden weinig ernstige bloedingen gezien rondom de PCI procedures, waarbij opgemerkt moet worden dat ongeveer de helft van de beschreven patiënten, milde hemofilie patiënten waren. De bloedingen die optraden tijdens en na PCI waren met name ter plaatse van de insteekopening.
Onder welke omstandigheden kunnen trombocyten aggregatieremmers veilig worden gegeven in het kader van secundaire preventie van coronaire events?
Aanbevelingen
Onderbouwing
Inleiding
Deze vraag betreft een situaties waar trombose al is opgetreden. De vraag naar het risico op coronaire events bij hemofilie is dus minder relevant. Voor deze vraagstelling wordt alleen naar het bloedingsrisico gekeken.
Conclusies
|
SORT Grade |
Conclusie |
|
B |
De frequentie van bloedingen onder aspirine lijkt wat groter dan in de algemene populatie, maar acceptabel in patiënten met milde hemofilie en in patiënten die stollingsfactorprofylaxe gebruiken. Er zijn weinig data in matig ernstige en ernstige hemofilie zonder stollingsfactorprofylaxe. (Boehnel, 2018; Schutgens, 2009) |
|
N.v.t. |
Er zijn onvoldoende data over clopidogrel monotherapie om een uitspraak te kunnen doen over bloedingsrisico. |
|
B |
Dubbele trombocytenaggregatieremming gedurende 1 maand lijkt veilig in patiënten met milde hemofilie, en in patiënten met ernstige of matig ernstige hemofilie die stollingsfactorprofylaxe gebruiken. Waarschijnlijk is het niet nodig om die stollingsfactorprofylaxe dagelijks te geven. Ervaring met langduriger dubbele trombocytenaggregatieremming is beperkt. (Boehnel, 2018) |
Samenvatting literatuur
Resultaten
Boehnel verrichtte een systematische review van PCI in patiënten met hemofilie , de meeste van hen werden behandeld met trombocytenaggregatieremming: Monotherapie met aspirine wordt beschreven in 38 patiënten: 15 milde, 9 matige (wv 3 met profylaxe) en 13 ernstige (wv 10 met profylaxe. Ernstige bloedingen traden op in 2/38 (1 mild, 1 matig zonder profylaxe). Milde bloedingen werden beschreven bij 4/38 (2 mild, 2 ernstig met profylaxe). Monotherapie met clopidogrel wordt beschreven in 4 patienten, steeds als alternatief voor dubbele trombocytenaggregatie remming; 1x nadat bloeding optrad onder dubbele trombocytenaggregatieremming). Twee van de vier patiënten hadden ernstige hemofilie, beiden zonder profylaxe. Er traden geen bloedingen op, waarbij opgemerkt moet worden dat de duur van behandeling maximaal 9 maanden was. Dubbele trombocytenaggregatieremming met clopidogrel plus aspirine wordt beschreven in 28 patiënten (9 mild (wv 2 op profylaxe), 6 matig (wv 3 op profylaxe), 12 ernstig (wv 9 op profylaxe) en 1 met onbekende ernst. Behandeling met dubbele trombocytenaggregatieremming werd gedurende maximaal 1 maand gegeven in 15/27 patiënten met bekende behandelduur. Bloedingen werden beschreven in 5/28 patiënten: 1 milde patiënt had minor bloeding, 1 matig ernstige patiënt zonder profylaxe had gewrichtsbloedingen. Alle drie de ernstige patiënten zonder profylaxe stopten dubbele trombocytenaggregatieremming vanwege bloedingen. Van de 15 patiënten op profylaxe spoten 5 dagelijks (2 ernstig, 1 matig, 2 mild). Van de milde patiënten waren er 2 ≥ 25%, 4 10-20% (incl de 2 op profylaxe), 2 < 10% en 1 onbekend (Boehnel, 2009).
Schutgens beschrijft, in a cross-sectionele Europese studie, 9 patiënten op monotherapie aspirine voor boezemfibrilleren (Schutgens, 2009). Tijdens gemiddeld 57 maanden follow-up werden geen bloedingscomplicaties gemeld bij 7 milde patiënten. Een ernstige en een matig ernstige patiënt hadden evenmin bloedingscomplicaties, maar gebruik van profylaxe werd niet beschreven.
Referenties
Bewijskracht literatuur
Level 2
Zoeken en selecteren
Er is een systematisch literatuursearch verricht in Pubmed (op 17-09-2018) met de volgende zoekstrategie:
(“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab])
AND
(“Platelet Aggregation Inhibitors” [Pharmacological Action] OR “Platelet Aggregation Inhibitors”[Mesh] OR “Anticoagulants”[Mesh] OR platelet aggregation inhib*[tiab] OR antiaggreg*[tiab] OR anti-aggreg*[tiab] OR platelet inhib*[tiab] OR antiplatelet*[tiab] OR anti-platelet*[tiab] OR platelet antagon*[tiab] OR anticoagula*[tiab] OR aspirin[tiab] OR clopidog*[tiab] OR ticagrel*[tiab] OR prasugrel*[tiab] OR dipyridamol*[tiab])
AND
(“Hemorrhage”[Mesh] OR hemorrhag*[tiab] OR haemorrhag*[tiab] OR bleeding[tiab])
De zoekstrategie leverde 502 hits op, waarvan 16 in fulltext beoordeeld zijn. Er werden geen interventiestudies gevonden. Verschillende artikelen beschreven single-centre ervaringen met gebruik van TAR. Een systematisch review vat deze data samen voor zover het gebruik na PCI betreft, voor december 2016 (Boehnel, 2017). Overige cohort studies of case-series werden gebruikt voor zover zij gebruik vanaf 2017 beschreven, of gebruik in een andere setting dan na PCI. Er werden verschillende review artikelen gevonden, waarvan 2 (van erkende experts) werden gebruikt voor de overwegingen.
Overwegingen
In het algemeen wordt ervan uitgegaan dat de indicatie voor trombocytenaggregatieremmers in patiënten met hemofilie na coronaire events dezelfde is als in de algemene populatie (Schutgens, 2009; Martin, 2016). Er zijn geen aanwijzingen dat patiënten met hemofilie bijvoorbeeld een lager risico op een recidief event zouden hebben. De meeste reviews beperken zich tot de cardiale indicaties. Het advies om na een acuut coronair syndroom zo mogelijk langdurig aspirine te gebruiken wordt steeds genoemd. Ook over het beleid na plaatsing van coronaire stents is consensus: zo mogelijk aspirine en clopidogrel, in ieder geval voor de minimale periode die de cardiale richtlijnen adviseren voor patiënten met een hoog bloedingsrisico. Een beperking van de beschikbare reviews is dat vaak dezelfde auteurs worden geciteerd, het gaat dus niet perse om onafhankelijke meningen.
De twee gebruikte reviews adviseren om bij ernstige hemofilie altijd stollingsfactorprofylaxe te geven voor monotherapie aspirine, en bij matig ernstige hemofilie stollingsfactorprofylaxe te overwegen (afhankelijk van vooral bloedingsfenotype). Bij milde hemofilie wordt suppletie niet nodig gevonden.
Bij combinatietherapie met aspirine en clopidogrel is er variatie in het beleid: Martin adviseert om stollingsfactorprofylaxe te geven met een dalspiegel van 10-15 IE/dl, Schutgens een dalspiegel van 30 IE/dl. Dat is (veel) intensiever dan veilig lijkt uit de literatuur. De meeste patiënten met milde hemofilie lijken dubbele plaatjesremming te verdragen zonder suppletie. Bij patiënten met ernstige en matig ernstige hemofilie is er consensus dat stollingsfactorprofylaxe nodig is, maar vaak lijkt het niet nodig om intensiever te suppleren dan met de reguliere stollingsfactorprofylaxe. In alle gevallen moet het individuele bloedingsfenotype worden meegewogen, en is frequent evaluatie met zo nodig aanpassen van de behandeling nodig.
Multidisciplinaire afweging van baten en risico van trombocytenaggregatieremming moet worden gemaakt door cardioloog en hemofiliebehandelaar, zo mogelijk vóor de keuze voor een bepaalde interventie wordt gemaakt (overig beleid rondom PCI wordt elders beschreven). Na start van trombocytenaggregatieremming, al dan niet met aanpassing van het profylactisch regime met stollingsfactor, moet de bloedingsfrequentie frequent worden geëvalueerd en het beleid zo nodig aangepast. Patiënten met hemofilie die een trombocytenaggregatieremmer gebruiken moeten daarbij altijd een protonpompremmer voorgeschreven krijgen.
Wanneer is medicamenteuze preventie van veneuze trombose aangewezen?
Aanbevelingen
Onderbouwing
Inleiding
Voor de vraag naar tromboseprofylaxe bij hemofilie patiënten zijn zowel het risico op veneuze trombo-embolie relevant als de bloedingsrisico’s van medicamenteuze behandeling.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
Spontane trombose komt slechts sporadisch voor bij patiënten met hemofilie. (Girolami, 2006) |
|
A |
De incidentie van trombose bij orthopedische chirurgie, grotendeels zonder medicamenteuze tromboseprofylaxe, varieert in de literatuur van 0% tot 4.3%. (Buckner, 2016; Raza, 2016; Perez Botero, 2015) |
|
A |
De incidentie van subklinische trombose bij orthopedische chirurgie, grotendeels zonder medicamenteuze tromboseprofylaxe, varieert in de literatuur van 0% tot 10%. (Buckner, 2016; Perez Botero, 2015; Hermans, 2010; Takedani, 2015) |
|
A |
Er is geen literatuur die aantoont dat post operatieve veneuze trombo-embolie vaker voorkomt bij patiënten met hemofilie B vergeleken met hemofilie A. (Buckner, 2016; Raza, 2016; Perez Botero, 2015; Hermans, 2010; Takedani, 2015) |
|
C |
Een voorgeschiedenis van trombose, een trombofilie factor, of een actieve maligniteit, kan een reden zijn om wel tromboseprofylaxe te geven bij patiënten met hemofilie A en B, zolang er sprake is van adequate substitutie van de stollingsfactordeficiëntie. (Ahmed, 2018) |
Samenvatting literatuur
Resultaten
Het vóórkomen van veneuze trombose
Over het algemeen wordt gedacht dat patiënten met hemofilie beschermd worden tegen veneuze tromboembolie door hun factor deficiëntie. Er zijn slechts enkele case reports over spontane trombose (Dargaud, 2003; Kashyao, 2006). Een review beschreef 27 patiënten met een niet-katheter gerelateerde trombose tot 2006. Bij 24 patiënten werd een risicofactor gevonden voor het ontstaan van de trombose. De meest voorkomende risicofactoren waren het ondergaan van een operatie, toediening van aPCC (FEIBA®) of rFVIIa (Novoseven®), een onderliggende trombofilie factor, maar ook toediening van FVIII of FIX (Girolami, 2006).
Een systematische review uit 2012 bekeek trombotische complicaties gerelateerd aan factor substitutie in patiënten met hemofilie in prospectieve studies. Er werden 71 studies geïncludeerd met in totaal 5528 patiënten met hemofilie A, hemofilie B en VWD. Binnen de hemofilie patiënten werden er 20 trombotische complicaties beschreven, waarvan 2 in hemofilie A patiënten (0,045%) en 11 in hemofilie B patiënten (1,47%). Dit waren 19 oppervlakkige tromboflebitiden ter plaatse van de infusie plek, bij 1 patiënt werd de aard van trombose niet beschreven. In de groep VWD patiënten werd, naast 5 oppervlakkige tromboflebitiden, 1 diep veneuze trombose en 1 longembolie beschreven bij patiënten die langdurige FVIII/VWF-concentraat kregen in verband met een operatie en waarbij er sprake was van zeer hoge FVIII waarden (248% resp. 450%) (Coppola, 2012).
Tijdens (orthopedische) chirurgie zou bescherming van de lage factor FVIII/FIX wegvallen door het toedienen van factorconcentraten. In een studie uit 1995 werd in patiënten zonder hemofilie, die geen operatie ondergingen, aangetoond dat een FVIII spiegel > 150% geassocieerd was met het ontwikkelen van een trombose (OR 4,8 95%Cl 2,3-10,0) (Hermans, 2009). Het is ook aangetoond dat de FVIII waarde in de eerste 24 uur na een operatie verdubbelt in patiënten zonder hemofilie (Hermanides, 2009). Echter in hemofilie A patiënten wordt de factor VIII stijging na substitutie voor een operatie nauwkeurig in de gaten gehouden en stijgt de factor VIII vaak niet zo hoog en langdurig als in patiënten zonder hemofilie. Dit geldt natuurlijk niet voor patiënten met hemofilie B. Er is geen literatuur die aantoont dat na gebruik van hooggezuiverd FIX-concentraat postoperatieve trombose vaker voorkomt bij patiënten met hemofilie B ten opzicht van hemofilie A.
Verschillende studies hebben gekeken naar het voorkómen van trombose tijdens orthopedische chirurgie. Een retrospectieve single center studie uit 2016 rapporteerde 18 patiënten met hemofilie A en 5 met hemofilie B die een totale heup of knieoperatie hadden ondergaan. Trombose profylaxe bestond uit intermitterende pneumatische compressie in 52%, enoxaparine in 4% en 43% van de patiënten kreeg geen trombose profylaxe. Na 1 jaar follow up werd er geen trombose beschreven (Raza, 2016). Een retrospectieve studie uit 2015 beschreef de eigen ervaring met 42 patiënten met hemofilie die in totaal 71 heup- of knieoperaties ondergingen en deed daarnaast een literatuurreview. Alle 42 patiënten kregen als tromboseprofylaxe steunkousen en 2 patiënten kregen LMWH profylaxe. Slechts 1 van de patiënten met een heupfractuur ontwikkelde een symptomatische DVT onder enoxaparine en bleek later ook heterozygoot voor factor V Leiden te zijn. In de literatuurreview werden in totaal 35 studies geïncludeerd, die 1170 procedures in 843 patiënten met hemofilie beschreven. Twee studies rapporteerden een incidentie van subklinische distale DVTs van resp. 0% en 10% (Hermans, 2010; Takedani, 1995). Met het meenemen van hun eigen resultaten kwamen zij uit op een incidentie van 0,5% voor klinische trombose en 0,25% voor subklinische trombose (Perez Botero, 2015) . Een prospectieve multicenter studie uit 2016 bekeek de prevalentie van trombose in 46 patiënten die een heup of knieoperatie ondergingen in 11 verschillende centra in Amerika. Bij de helft van de patiënten was er sprake van tromboseprofylaxe in de vorm van IPC, 20% kreeg steunkousen en 4 patiënten kregen LMWH. Bij alle patiënten werd diagnostiek gedaan naar distale diep veneuze trombose 4-6 weken na de operatie. 1 patiënt met milde hemofilie A ontwikkelde een distale diep veneuze trombose, 1 patiënt met ernstige hemofilie A ontwikkelde een longembolie, bij deze patiënt was er sprake van een FVIII piekwaarde van >200%. Geen van de patiënten ontwikkelde een subklinische DVT. Prevalentie van DVT in deze studie was 4,3% (95%CI 0,5-14,8%) (Buckner, 2016). In deze studie was er sprake van een majeure bloeding, gedefinieerd als een hemoglobine daling van > 2 punten in 24 uur of transfusie noodzaak bij 18 (40%) patiënten, waaronder 2 van de 4 patiënten die profylactisch LMWH kreeg.
Tromboseprofylaxe
Er zijn verschillende internationale surveys gedaan naar hoe tromboseprofylaxe bij PWH in de dagelijkse praktijk wordt toegepast. Een multicenter Europese survey vond dat 50% van de 26 centra trombose profylaxe gaven bij grote orthopedische operaties (Hermans, 2009). Een korte survey onder 140 hemofilie behandel centra in Amerika liet zien dat 67% van de respondenten zouden kiezen voor tromboseprofylaxe in alle patiënten met hemofilie die een heup of knie operatie ondergaan. Van de respondenten die niet bij alle patiënten tromboseprofylaxe zouden geven, zou 78% wel tromboseprofylaxe geven in een geselecteerde groep, bijvoorbeeld patiënten die na substitutie een factorspiegel > 100 IE/dL hadden. De manier van tromboseprofylaxe was steunkousen bij 32%, 24% LMWH, 1% fondaparinux, 3% ongefractioneerde heparine, 4% warfarine en 1% aspirine (Pradhan, 2009).
In 2018 is er een Europese richtlijn verschenen over peri-operatieve tromboseprofylaxe. Hierin is een apart stuk opgenomen over tromboseprofylaxe bij patiënten met een bloedingsziekte. Drie hematologen en 2 anesthesisten hebben de literatuur onderzocht en aanbevelingen gegeven. Zij adviseerden het volgende: “bepaal per patiënt het tromboserisico op basis van de aard van de ingreep en operatie, voorgeschiedenis van trombose of maligniteit, ernst van de hemofilie en bloedingsfenotype; vermijd het toedienen van teveel stollingsfactor en monitor de factorlevels nauwkeurig, vermijd FVIII levels > 150 IE/dl; pas mechanische tromboseprofylaxe in de vorm van steunkousen toe; geef geen routinematig tromboseprofylaxe aan patiënten met hemofilie A of B; als er gekozen wordt voor medicamenteuze tromboseprofylaxe, kies dan voor LMWH onder adequate factorsubstitutie (Ahmed, 2018)”.
De Nederlandse richtlijn antitrombotisch beleid uit 2016 geeft bij medicamenteuze tromboseprofylaxe ook de voorkeur aan LMWH.
Referenties
Bewijskracht literatuur
Level 3
Zoeken en selecteren
20-09-2018 Pubmed: (((“acute coronary syndrome”[Mesh] OR “Percutaneous coronary intervention”[MESH]))) AND ((“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab])). Aantal hits: 33
20-09-2018 Pubmed: (“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab]) AND (“Anticoagulation” [Pharmacological Action] OR “Anticoagulation” [Mesh] OR “Anticoagulants”[Mesh] OR anticoagu*[tiab] OR antico*[tiab] OR anti-coagu*[tiab] OR anticoagula*[tiab] OR heparin[tiab] OR low molecular weight*[tiab] OR oral anticoagul*[tiab] OR LMWH*[tiab] OR bivalirudin*[tiab] OR VKA*[tiab] OR vitamin K antagonists*[tiab])
AND (“Hemorrhage”[Mesh] OR hemorrhag*[tiab] OR haemorrhag*[tiab] OR bleeding[tiab]). Aantal hits: 478.
Vanuit de Pubmed strategie en vanuit de referentielijsten van de geselecteerde artikelen of vanuit de richtlijnen werden in totaal 14 full text artikelen geselecteerd voor deze uitgangsvraag.
Daarnaast werden de Nederlandse richtlijn Antitrombotisch beleid van de Nederlandse Internisten Vereniging en de Richtlijn voor de chirurgische behandeling van hartklep-aandoeningen van de Nederlandse Vereniging voor Thoraxchirurgie geraadpleegd.
Overwegingen
Over het algemeen wordt gedacht dat patiënten met hemofilie beschermd worden tegen veneuze tromboembolie door hun factor deficiëntie. De incidentie van spontane trombose in hemofilie patiënten is heel laag. Bij patiënten die een orthopedische operatie en stollingsfactorsubstitutie krijgen, vervalt deze bescherming tegen trombose. Niettemin blijft de incidentie van trombose na een operatie laag, variërend in de literatuur van 0% tot 4,3% voor klinische trombose en 0% tot 10% voor subklinische trombose, waarbij er bij de meeste patiënten geen medicamenteuze tromboseprofylaxe gegeven is. Als meest waarschijnlijke verklaring wordt genoemd dat de FVIII bij hemofilie A patiënten die stollingsfactorsubstitutie krijgen, nooit zo hoog stijgt als de factor FVIII in de algemene populatie tijdens een operatie. Enkele casus beschrijven trombose in hemofilie en VWD patiënten waarbij er sprake was van een FVIII > 200 IE/dL tijdens substitutie therapie. In de algemene populatie is het hebben van een FVIII > 150 IE/dL geassocieerd met het ontwikkelen van een trombose. Bij het geven van stollingsfactorsubstitutie moet dus gewaakt worden voor te hoge FVIII spiegels.
Onder welke omstandigheden kunnen anticoagulantia veilig worden gegeven bij de therapeutische behandeling van reeds opgetreden trombose in patiënten met hemofilie?
Aanbevelingen
Onderbouwing
Inleiding
Deze vraag betreft een situaties waar trombose al is opgetreden. De vraag naar het risico op veneuze trombo-embolie bij hemofilie is dus minder relevant. Voor deze vraagstelling wordt alleen naar het bloedingsrisico gekeken.
Conclusies
|
SORT Grade |
Conclusie |
|
A |
Het korter dan drie maanden behandelen van een diep veneuze trombose leidt tot verhoogd risico op recidief trombose in de algemene populatie. (Schulman, 1995) |
|
N.v.t. |
Het gehalte stollingsfactor waarbij therapeutische antistolling veilig kan plaatsvinden is in de literatuur niet uitgezocht. |
|
A |
Het gebruik van DOACS in patiënten zonder hemofilie is geassocieerd is met minder intracraniële bloedingen en minder fatale bloedingen ten opzichte van VKA. (Es, 2014; richtlijn antitrombotisch beleid, 2016) |
|
N.v.t. |
Er is geen ervaring met DOACs in de behandeling van veneuze tromboembolie bij patiënten met hemofilie. |
Samenvatting literatuur
Resultaten
De behandeling van diep veneuze trombose in patiënten met hemofilie is niet in studies uitgezocht. Uit de beschreven case reports blijkt dat de behandeling van DVT erg varieert (Buckner, 2016; Bicer, 2009; Ettingshausen, 1999) en soms geassocieerd is met bloedingen (Ritchie, 1992). In Nederland wordt een diep veneuze trombose behandeld met directe orale antistollings middelen (DOACs) of LMWH in combinatie met vitamine K antagonisten (VKA) voor tenminste 3 maanden. Er zijn aanwijzingen dat het gebruik van DOACS in patiënten zonder hemofilie geassocieerd is met minder intracraniële bloedingen ten opzichte van VKA (RR 0,37 95%CI 0,21-0,68) en minder fatale bloedingen (RR 0,36 95%CI 0,15-0,84) (Es, 2014; richtlijn antitrombotisch beleid, 2016). Echter, er is geen ervaring met DOACS bij patiënten met hemofilie.
Het gehalte stollingsfactor waarbij therapeutische antistolling veilig kan plaatsvinden is in de literatuur niet uitgezocht. Expert opinion in eerdere artikelen neigt naar 30 IE/dL, terwijl in meer recente literatuur zowel 20 als 30 IE/dL wordt genoemd (Schutgens, 2016; Martin, 2016).
Martin et al. adviseert een behandelduur van 1 maand, omdat in de algemene populatie na 1 maand het risico op trombose recidief flink daalt (cumulatieve incidentie na 30 dagen 5,2%, na 90 dagen 8,3%) (Martin, 2016; Heit, 2000). Echter de DURAC studie van Schulman et al. laat zien dat trombose behandeling korter dan 3 maanden leidt tot een hogere recidief kans in de algemene populatie (Schulman, 1995).
Referenties
Bewijskracht literatuur
Level 3
Zoeken en selceteren
20-09-2018 Pubmed: (((“acute coronary syndrome”[Mesh] OR “Percutaneous coronary intervention”[MESH]))) AND ((“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab])). Aantal hits: 33
20-09-2018 Pubmed: (“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab]) AND (“Anticoagulation” [Pharmacological Action] OR “Anticoagulation” [Mesh] OR “Anticoagulants”[Mesh] OR anticoagu*[tiab] OR antico*[tiab] OR anti-coagu*[tiab] OR anticoagula*[tiab] OR heparin[tiab] OR low molecular weight*[tiab] OR oral anticoagul*[tiab] OR LMWH*[tiab] OR bivalirudin*[tiab] OR VKA*[tiab] OR vitamin K antagonists*[tiab])
AND (“Hemorrhage”[Mesh] OR hemorrhag*[tiab] OR haemorrhag*[tiab] OR bleeding[tiab]). Aantal hits: 478.
Vanuit de Pubmed strategie en vanuit de referentielijsten van de geselecteerde artikelen of vanuit de richtlijnen werden in totaal 9 full text artikelen geselecteerd voor deze uitgangsvraag.
Daarnaast werd de Nederlandse richtlijn Antitrombotisch beleid van de Nederlandse Internisten Vereniging geraadpleegd.
Overwegingen
De behandeling van diep veneuze trombose in hemofilie is niet uitgezocht, in de beschreven casuïstiek wordt er verschillend beleid gevoerd. In Nederland is de behandeling van trombose beschreven in de richtlijn Antitrombotisch beleid en wordt een diep veneuze trombose behandeld met DOACs of LMWH en VKA voor tenminste 3 maanden. Er zijn aanwijzingen dat het gebruik van DOACs in patiënten zonder hemofilie geassocieerd is met minder bloedingscomplicaties ten opzichte van VKA. Gebruik van VKA geeft een onvoorspelbare (en niet meegenomen in monitoring middels INR) daling van factor IX. Daarmee zijn VKA moeilijk toepasbaar in hemofilie B.
Over het algemeen wordt aangehouden dat voor het geven van therapeutische antistolling (VKA of LMWH) een dalspiegel van factor VIII/IX > 30-20 IE/dL nagestreefd wordt. In de praktijk blijkt 30 IE/dL niet haalbaar voor patiënten met ernstige hemofilie. Daarom adviseert deze richtlijn een dalspiegel > 20 IE/dL. In de algemene populatie wordt geadviseerd een trombose ten minste 3 maanden te behandelen en korter behandelen leidt tot een hogere recidief kans in de algemene populatie. Derhalve sluiten wij aan bij de antitrombotische richtlijn die ten minste 3 maanden antistolling adviseert voor de behandeling van diep veneuze trombose.
Wanneer zijn anticoagulantia aangewezen ter voorkoming van herseninfarct bij boezemfibrilleren?
Aanbeveling
Onderbouwing
Inleiding
Om deze vraag te kunnen beantwoorden is zowel het risico op herseninfarct bij hemofilie patiënten met boezemfibrilleren relevant als het bloedingsrisico van antistolling. De nadruk in dit deel ligt op het risico op herseninfarct. Het bloedingsrisico van antistolling is eerder uitgewerkt, in de vraag over veneuze tromboembolie.
Conclusie
|
SORT Grade |
Conclusie |
|
N.v.t. |
Het is niet bekend hoe groot het risico op herseninfarct is in hemofilie patiënten met atriumfibrilleren. |
Samenvatting literatuur
Resultaten
Schutgens beschrijft gemiddeld 57 maanden follow-up in 33 patiënten met AF, zonder trombotische complicaties (2 patiënten op VKA, 9 patiënten op aspirine). Twee patiënten maakten een stroke door voor AF werd vastgesteld: bij een was AF op dat moment uitgesloten, voor de andere was die informatie niet beschikbaar (Schutgens, 2014). Humphries beschrijft 4 pat met AF, van wie er één een stroke heeft doorgemaakt. Er zijn geen details over ernst van hemofilie en aard van stroke (Humphries, 2015).
Referenties
Bewijskracht literatuur
Level 3
Zoeken en selecteren
Er is een systematisch literatuursearch verricht in Pubmed (op 17-09-2018) met de volgende zoekstrategie:
(“Hemophilia A”[Mesh] OR “Hemophilia B”[Mesh] OR hemophil*[tiab] OR haemophil*[tiab])
AND
(“Atrial Fibrillation”[Mesh] OR atrial fibrillat*[tiab])
De zoekstrategie leverde 35 hits op, waarvan 13 in fulltext beoordeeld zijn. Er werden twee cohortstudies gevonden die het vóorkomen van herseninfarct bij AF beschrijven. Een aantal andere studies was bruikbaar voor de overwegingen.
Overwegingen
Boezemfibrilleren komt bij hemofilie even vaak voor als in de algemene populatie (Martin, 2016; Humphries, 2015; Minuk, 2015; Schutgens, 2014). In een cross-sectionele Europese studie werd een prevalentie van 3,4% gezien in patiënten ouder dan 60 jaar (Schutgens, 2014). Behandelaren schatten het risico op herseninfarct bij AF kleiner in dan in de algemene populatie, maar data ontbreken. Het aantal patiënten met hemofilie en AF dat in de literatuur is beschreven is te klein om een harde uitspraak te doen. Indirect bewijs komt uit metingen van trombinegeneratie.
Om iets te kunnen zeggen over de mate waarin hemofilie patiënten in staat zijn stolsels te maken bij AF, is trombinegeneratie in hen vergeleken met die in gebruikers van vitamine K antagonisten. De Koning beschrijft dat trombinegeneratie in ernstige hemofilie vergelijkbaar is met die in VKA gebruikers. In patiënten met een factor VIII tussen de 1 en 19 IE/dl is de trombinegeneratie vergelijkbaar met VKA gebruikers met een INR van 1,5-1,9. Bij factor VIII spiegels van 20-50IE/dl stijgt de trombinegeneratie verder, tot aan de onderkant van het bereik in gezonde controles (de Koning, 2017). Königsbrügge rapporteert dat de gemiddelde trombinegeneratie in patiënten met ernstige hemofilie A (deels met aantoonbaar exogeen factor VIII) lager is dan in een groep van patiënten zonder hemofilie met AF die diverse antistollingsmiddelen gebruiken (Konigsbrugge, 2017).
In de algemene bevolking wordt geen aspirine gegeven voor AF, omdat de afweging tussen baten en schade van dit middel ongunstiger uitvallen dan voor antistolling. Er zijn geen data die ondersteunen dat dit anders is voor patiënten met hemofilie .
Praktische leidraad voor de follow-up, patiënten voorlichting & educatie, en zelfmanagement van patiënten met een bloedstollingsstoornis.
De werkgroep bestaande uit leden van de NVHP, NVHV en NVHB, vindt dat voorlichting en educatie van patiënten met een bloedstollingsstoornis van groot belang is om optimale zelfmanagement en therapietrouw te bewerkstelligen zodat de kwaliteit van korte en lange termijn uitkomsten geborgd zijn. Op verschillende manieren wordt in dit document aangegeven hoe (of de wijze waarop) zelfmanagement kan worden bevorderd. De regievoering hierin verschilt per Hemofiliebehandel-centrum. Er is getracht veel voorkomende vragen te beantwoorden en mogelijke knelpunten in de zorg te identificeren.
Er is geen literatuuronderzoek verricht voor deze uitgangsvraag, omdat vorm en inhoud van de zorg afhankelijk zijn, ons inziens, van de context waar binnen deze zorg geleverd wordt. We hebben ons daarom gebaseerd op de “expert opinion” van de werkgroep, in nauwe samenwerking met patiënten, luisterend naar en gebruikmakend van hun ervaringen.
Na een korte inleiding over de organisatie van de zorg voor patiënten met een bloedstollingsstoornis in Nederland, is getracht om puntsgewijs, in een zo logisch mogelijke volgorde richtlijnen en adviezen voor deze patiëntengroep te benoemen. Uiteraard worden bij elk patiëntencontact, door de contactpersoon belangrijke zaken herhaald. In deze module wordt echter verondersteld dat eerder vermelde kennis bekend is, om herhalingen te voorkomen en de leesbaarheid te vergroten. De stuurgroep beseft dat dit hoofdstuk nooit compleet kan zijn, maar hoopt hiermee wel tot steun te zijn voor patiënten, hun families en behandelaren van patiënten met een bloedstollingsstoornis.
Organisatie Hemofiliebehandelcentra
In Nederland is de zorg voor alle patiënten met een bloedstollingsstoornis gecentraliseerd. Deze gespecialiseerde zorg vindt plaats in zes door het Harmonisatie Kwaliteitsbeoordeling in de Zorgsector (HKZ) geaccrediteerde Hemofiliebehandelcentra waarin zowel kinderen als volwassenen worden behandeld. De details t.a.v. de organisatie van deze zorg, verschillen per behandelcentrum maar de organisatie is in grote lijnen vergelijkbaar. Elk hemofiliebehandelteam bestaat uit gespecialiseerde medisch specialisten (internist-hematologen, internist-vasculair geneeskundigen, kinderarts-hematologen), verpleegkundig specialisten, gespecialiseerde verpleegkundigen, maar ook een gespecialiseerde fysiotherapeut, een medisch maatschappelijk werker en eventueel een psycholoog. Het behandelteam werkt nauw samen met andere specialisten (vanuit o.a. orthopedie, revalidatie, infectieziekten, gynaecologie, anesthesie, maag-darm en leverziekten) en paramedici in- en buiten het ziekenhuis om de beste zorg voor elke patiënt vorm te geven.
Regievoering
Bij elke fase moet duidelijk zijn wie bij welk soort vraag het aanspreekpunt is voor de patiënt. Ieder Hemofiliebehandelcentrum bepaalt wie verantwoordelijk is voor welk deel van de voorlichting, dit wordt gedeeld met de patiënt zodat deze weet bij wie hij een vraag het best neer kan leggen.
Samen beslissen of “shared decision making”
Beslissingen worden zoveel mogelijk genomen samen met de patiënt, dit heet ook wel “shared decision making”. Uiteraard kan het voorkomen dat meningen niet overeen komen, dan zal in sommige gevallen het medisch inzicht de doorslag geven.
Gebruik
In de uitwerking van dit hoofdstuk komen verschillende stadia in het ziektebeloop & diverse levensfasen aan bod waarbij relevante informatie voor die fase wordt besproken.
Algemeen t.a.v. behandeling/ “Do’s”
Algemeen t.a.v. behandeling/ “Don’ts”
Reizen naar & verblijf in het buitenland
Naast algemene leefregels ook:
A. Rol klinisch geneticus binnen het Hemofiliebehandelcentrum
B. Aandachtspunten vanuit het behandelteam m.b.t. erfelijkheid, kinderwens, zwangerschap & bevalling
A. Algemeen
Aandoening gerichte informatie
Uiteraard worden accenten bij bovenstaande punten anders gelegd wanneer het andere categorieën patiënten wat betreft o.a. ernst van de ziekte, leeftijd, levensfase of andere bijzonderheden.
B. Aandachtspunten voor specifieke groepen zoals: het jonge kind en de ouders, mannelijke patiënten, vrouwelijke patiënten en draagsters van hemofilie
Voor vrouwen is aandacht met name van belang m.b.t.:
En specifiek bij draagsters:
A. Start profylaxe bij ernstige bloedstollingsstoornissen; uitgangspunt jonge kinderen (0-4 jaar)
Patiënt/ ouders of voogd gericht
Aandoening gericht
Leeftijd en levensstijlgericht
B. Toenemende zelfstandigheid van het groeiende kind (>4 jaar)
Patiënt/ ouders of voogd gericht
Aandoening gericht
Leeftijd en levensstijl gericht
C. Aandacht toenemende capaciteit tot zelf management (>8-10 jaar)
Patiënt/ ouders of voogd gericht
Aandoening gericht
Leeftijd en levensstijl gericht
Patiënt/ouders of voogd gericht
Aandoening gericht
Leeftijd en levensstijl gericht
A. Werken aan transitie tot jong volwassene, en volwassen zorg (>10-18 jaar)
B. Additionele aandachtspunten t.a.v. jong volwassenen (±18-25 jaar)
Specifieke aandacht voor reizen naar en verblijf in het buitenland
Patiënt gericht
Aandoening gericht
Leeftijd en levensstijl gericht
Kinderwens & zwangerschap
Reizen/buitenland (actieve vakanties)
Patiënt gericht
Aandoening gericht
Leeftijd en levensstijl gericht
Reizen/buitenland (actieve of minder actieve vakanties)
Patiënt gericht
Aandoening gericht
Leeftijd & levensstijl gericht
Reizen/buitenland (overwinteren)
Belangrijke links / informatie
© 2022. Alle rechten voorbehouden
Nederlandse Vereniging voor Hematologie (NVvH)
Nederlandse Vereniging voor Kindergeneeskunde (NVK)
Nederlandse Vereniging van Internisten Vasculaire Geneeskunde (NVIVG)
Nederlandse Vereniging voor Obstetrie en Gynaecologie (NVOG)
Nederlandse Orthopaedische Vereniging (NOV)
Vereniging Klinische Genetica Nederland (VKGN)
Nederlandse Vereniging Hemofilie Verpleegkundigen (NVHV)