Aanbevelingen
Zie ook MDS Europe – Patient management (mds-europe.org)
Tabel 1
|
Anamnese |
Klachten samenhangend met cytopenie, inspanningsintolerantie. Koorts, nachtzweten, gewrichtspijnen, huidafwijkingen. Toxische blootstelling in voorgeschiedenis: behandeling met chemotherapie/bestraling, benzeen, alcohol, (langdurige) behandeling met cytotoxische medicatie zoals methotrexaat Familieanamnese gericht op voorkomen van hematologische ziekten of solide tumoren geassocieerd met erfelijk voorkomen. |
|
Lichamelijk onderzoek |
‘WHO-performance status’. Huidafwijkingen (bijvoorbeeld ‘Sweet’ syndroom, perichondritis, pyoderma gangrenosum, vasculitis of huidlokalisatie). Vroegtijdig grijs (<30 jaar), nageldystrofie. |
|
Aanvullend onderzoek in perifeer bloed |
Hemoglobine (Hb), MCV, trombocyten, leukocyten, neutrofielen getal, leukocyten differentiatie met microscopie (dysplasie, circulerende blasten), reticulocyten, haptoglobine, directe antiglobuline test.
Foliumzuur, vitamine B12 IJzer, ferritine, TYBC, ijzerverzadiging
Leverenzymwaarden, LDH kreatinine, urinezuur, TSH, (FT4)
Immuunfenotypering: PNH bij aanwijzingen voor hypoplastische MDS, hemolyse en als verlies van CD14 op monocyten en/of CD16 op granulocyten bij immuunfenotypering van beenmerg. De monocyten-assay bij monocytose en verdenking CMML waarbij het percentage ‘classical monocytes’ wordt bepaald.2
Virus serologie (Parvo B19), HIV, CMV, Hepatitis B/C
Endogene EPO spiegel (in ieder geval bij anemie met behandelindicatie)
Bezinking en CRP (VEXAS en auto-inflammatoire beelden bij MDS, ANA en anti-ds DNA op indicatie) UBA1 mutatie bij verdenking VEXAS |
|
Beenmergonderzoek |
Aspiraat voor 1. Cytologie: cellulariteit, evaluatie dysplasie in 1 of meer cellijnen, ijzerkleuring voor ringsideroblasten, % blasten 2. Cytogenetica: detectie van chromosomale afwijkingen door middel van bijvoorbeeld karyotypering, FISH met geselecteerde ‘MDS’-probes en aantonen van CNLOH op 17p Onderzoeken die kunnen bijdragen aan het stellen van de diagnose 3. NGS/mutatieanalyse: detectie van somatische mutaties, uitgevoerd volgens aanbevelingen van de IPSS-M3. UBA1 mutatie bij verdenking VEXAS. 4. Immuunfenotypering: detectie van abnormale voorloper cellen en afwijkende expressie van differentiatie antigenen in myelo-monocytaire en erythroïde reeks, uitgevoerd volgens de European Leukemia Net (ELN) richtlijnen4 5. Biopt voor histologie en immuunhistochemie: voor cellulariteit, aantonen van dysplasie van met name megakaryocyten, % CD34+-blasten en abnormale lokalisatie en clustering, mate van fibrose |
Onderbouwing
Van een relevante cytopenie wordt gesproken bij de volgende waarden:5,6
De diagnose MDS wordt nogal eens gesteld door het bij toeval vinden van een afwijkend bloedbeeld, terwijl de patiënt nagenoeg geen klachten heeft. Symptomatologie hangt nauw samen met de cytopenie in één of meer cellijnen. Mogelijke klachten zijn: vermoeidheid en kortademigheid door anemie, recidiverende (bacteriële) infecties door neutropenie (aanwezig bij 50% van de patiënten bij diagnose) en/of granulocytaire/monocytaire dysfunctie en een verhoogde bloedingsneiging van huid en slijmvliezen door trombopenie (aanwezig bij 25% van de patiënten bij diagnose). Ook is er regelmatig sprake van een trombopathie waardoor ondanks een normaal of laag normaal trombocytengetal sprake kan zijn van een verhoogde bloedingsneiging. MDS is vaak geassocieerd (20-50%) met auto-immuun fenomenen, zoals artritis, polychondritis, spierpijn, vasculitis en huidafwijkingen.7 Bij auto-inflammatoire beelden is het belangrijk te denken aan het VEXAS-syndroom.8
Over het algemeen kan de diagnose zonder al te grote problemen worden gesteld wanneer het een oudere patiënt betreft met een pancytopenie en trilineaire dysplasie in bloed en/of beenmerg. Dysplasie is niet specifiek voor MDS maar kan men ook zien bij megaloblastaire anemie, overmatig alcoholgebruik, infectieziekten (denk aan HIV, TBC), hemolyse en tijdens of na cytostaticagebruik. (Tabel 2) Andere oorzaken van cytopenie(ën) moeten worden uitgesloten. De diagnostiek is ingewikkelder bij patiënten met één of meerdere cytopenie(ën) waarbij het beenmerg amper dysplastisch is, zonder toename van blasten en zonder specifieke cytogenetische of moleculaire afwijkingen (ICUS: idiopathic cytopenia of undetermined significance; CCUS: clonal cytopenia of undertemined significance zie ook later). In deze situatie kan een flow cytometrische analyse bijdragen aan het objectiveren van dysplasie.9,10 Soms wordt de dysplasie pas echt duidelijk na herhaald beenmerg onderzoek. Bij een hypoplastisch beenmerg kan het onderscheid met aplastische anemie of paroxysmale nachtelijke hemoglobinurie (PNH) moeilijk zijn.
Tabel 211
|
Differentiaal diagnose van cytopenieën en/of dysplasie |
Laboratorium onderzoek |
|
Vitamine deficiëntie |
Vitamine B12, foliumzuur |
|
Toxische schade (alcohol, lood, zink, koper deficiëntie, chemotherapie) |
Lood, zink, koper |
|
Reactieve veranderingen als gevolg van (chronische) inflammatie (infectie, HIV, hepatitis, systeemziekten, VEXAS) |
HIV, hepatitis, Parvo B19 |
|
Aplastische anemie, ‘pure red cell aplasia’, paroxysmale nachtelijke hemoglobinurie |
Beenmergonderzoek met immuunhistochemie, immuunfenotypering op perifeer bloed voor verlies van glycosylphophatidylinositol (GPI)-geankerde eiwitten |
|
Medicamenteus (bijvoorbeeld methotrexaat, azathioprine, valproïnezuur, levetiracetam, ganciclovir, sulfonamiden, colchicine, NSAID’s) |
|
|
ICUS/CCUS/IDUS/CHIP |
Beenmergonderzoek met morfologie, immuunfenotypering en histologie met immuunhistochemie, cytogenetica en NGS |
|
Andere hematologische ziekte (acute leukemie (ALL, AML), large granular lymphocytic leukemia, myeloproliferatieve ziekte: CMML, MDS/MPN met neutrofilie, primaire myelofibrose) |
Beenmergonderzoek met morfologie, immuunfenotypering en histologie en immuunhistochemie, cytogenetica (en NGS) |
|
Congenitale vorm van beenmerg falen: ‘congenital dyserythropoietic anemia’ |
NGS gericht op beenmerg falen |
|
Immuun trombocytopenie |
Autoantistoffen tegen trombocyten |
|
Beenmerglokalisatie van een solide maligniteit |
Beenmergonderzoek histologie en immuunhistochemie |
Diagnostiek
De diagnose van MDS is gebaseerd op (cyto)morfologisch onderzoek van het bloed en beenmerg. Bijkomende diagnostische en prognostische informatie wordt verkregen uit cytogenetisch, moleculair en histopathologisch onderzoek en het beenmergbiopt. In figuur 1 is het diagnostisch stroomdiagram weergegeven voor de diagnostiek van cytopenie(ën) en bij verdenking op MDS. Het is een pragmatische benadering omdat de doorlooptijd van de afzonderlijke onderzoeken verschillend is. Zo is de morfologie binnen enkele uren te beoordelen en kan het inzetten van het vervolgonderzoek richting geven. Immuunfenotypering is doorgaans binnen enkele uren tot 24 uur bekend. Afhankelijk van de bevindingen van cytomorfologisch onderzoek (en immuunfenotypering) kan besloten worden cytogenetica en NGS in te zetten. Het wordt aanbevolen om beenmerg in te vriezen zodat in een later stadium alsnog besloten kan worden om NGS of onderzoek naar translocaties in geval van MDS/AML in te zetten. De doorlooptijd van cytogenetica en NGS is meestal 1-4 weken. Het is belangrijk om de afzonderlijke uitslagen integraal te bespreken en te verslaan in een rapport voor het stellen van de diagnose.
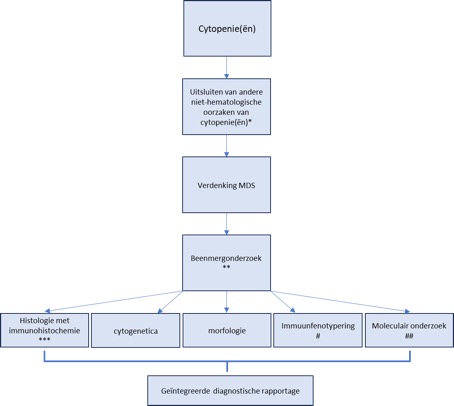
Figuur 1 toont een stroomdiagram van de diagnostische benadering van patiënten met een cytopenie en verdenking op MDS. Hierin zijn de belangrijkste ontwikkelingen samengevat. (aangepast naar van de Loosdrecht et al, Cytometry part B: Clinical Cytometry 2023)12
*zie Tabel 1.
** ook ter uitsluiting van andere hematologische oorzaken van cytopenie(ën).
*** moleculair-genetisch onderzoek op het beenmergbiopt is optioneel wanneer het niet mogelijk is om dit op beenmerg of perifeer bloed te verkrijgen. Bij patiënten met MDS en toename blasten in het beenmerg, maar niet in het perifeer bloed is het aan te bevelen om moleculair onderzoek op beenmerg te verrichten.
# immuunfenotypering kan helpen bij het stellen van de diagnose MDS.
## moleculair onderzoek middels NGS wordt aanbevolen.
Morfologie van bloed en beenmerg
Er is pas sprake van dysplasie in één of meer cellijnen indien in tenminste 10% van de cellen in een cellijn dysplastische kenmerken worden gezien.5,6 Tabel 3 toont de meest voorkomende cytologische afwijkingen en dysplastische kenmerken in het bloed en beenmerg bij patiënten met MDS. Bij een MDS met een laag MCV zonder ijzergebrek, wat in zeldzame gevallen voorkomt, kan sprake zijn van een verworven alfa-thalassemie op basis van een ATRX-mutatie.13 Het percentage blasten dat gebruikt wordt in de huidige diagnostische en prognostische classificaties is gebaseerd op cytomorfologie. Alleen als het aspiraat van slechte kwaliteit is door bijvoorbeeld ziekte specifieke redenen (dry tap) kan het percentage blasten bij beoordeling van het biopt worden geschat. De beoordeling van dysmegakaryopoïese kan lastig zijn in een cytologisch preparaat, met name ook omdat het aantal megakaryocyten bij MDS meestal laag is.
In de WHO 2022 en ICC classificatie voor MDS is de nadruk meer komen te liggen op het identificeren van subgroepen op basis van genetische en moleculaire afwijkingen in tegenstelling tot de voorgaande WHO classificaties.5,6 Dysplasie is geen voorwaarde meer om de diagnose MDS te stellen bij aanwezigheid van cytopenie(en) met een SF3B1 mutatie of deletie 7/7q, complex afwijkend karyotype of multihit TP53 mutatie.6De aanwezigheid van unilineaire of multilineaire dysplasie wordt als onderscheidend voor MDS NOS met unilineaire en multilineaire dysplasie beschouwd in de ICC, maar binnen de WHO 2022 niet meer. 5,6
Tabel 3 veel voorkomende cytologische afwijkingen en dysplastische kenmerken bij MDS5
|
Dyserythropoëse |
|
|
Kern |
‘budding’ |
|
|
Internucleaire bruggen |
|
|
meerkernigheid |
|
|
megaloblastaire veranderingen |
|
|
karyorrhexis |
|
Cytoplasma |
ring sideroblasten |
|
|
vacuolisatie |
|
|
positieve ‘periodic acidic schiff’ (PAS) kleuring |
|
Dysgranulopoiese |
|
|
Kern |
hyposegmentatie (pseudo-Pelger-Huët) |
|
|
hypersegmentatie |
|
Cytoplasma |
hypogranulatie |
|
|
Pseudo-Chédiak-Higashi granules |
|
kleine kern |
|
|
|
Auerse staven |
|
Dysmegakaryopoiese |
|
|
Kern |
hypolobulatie |
|
|
meerkernigheid |
|
Cytoplasma |
micromegakaryocyten (kan een aanwijzing zijn voor MDS met deletie 5q) |
Histopathologisch en immuunhistochemisch onderzoek van het beenmergbiopt
Tenslotte is ook immuunhistochemisch (IHC) onderzoek op een beenmergbiopt van belang.14 Een minimale set aan markers is gedefinieerd en omvat markers als CD34, CD117 (progenitor cellen), MPO (myelo-[monocytair]), CD14, en/of CD68 (monocyten/macrofagen), glycophorine-C (rode bloedcelvoorloper cellen), CD61 en CD42 (megakaryocytair). Deze immuunhistochemische kleuringen dragen bij aan het beter onderscheiden van de subpopulaties in het beenmerg ten aanzien van frequentie van voorkomen en aanwezigheid van dysplasie. Een biopt geeft ook goede informatie over cellulariteit van het beenmerg en de mate van fibrose die gepaard kan gaan met een MDS. De mate van fibrose lijkt van belang als prognostische factor en wordt in de WHO 2022 erkent als een aparte subgroep van MDS.5,15 De mate van fibrose in een beenmergbiopt van focaal tot uitgebreid is nog weinig gestandaardiseerd.
Cytogenetisch onderzoek
Bij 40 tot 80% van de patiënten met een MDS zijn specifieke chromosomale afwijkingen te vinden, zoals bijvoorbeeld deletie van een deel van de lange arm van chromosoom 5 (de zogenaamde del(5q)), en het ontbreken van één chromosoom 7 (monosomie 7). Gebalanceerde translocaties die men typisch bij (jonge) AML-patiënten aantreft, komen ook voor bij MDS, maar zijn relatief zeldzaam bij MDS. Bij patiënten die MDS ontwikkeld hebben na vroegere blootstelling aan cytostatica of bestraling (de zogenoemde therapie-gerelateerde MDS) worden vaak complexe (³ 3) cytogenetische afwijkingen gevonden. Afwijkingen in het karyotype geven niet alleen belangrijke diagnostische, maar ook prognostische informatie (zie verder). Wanneer met standaardtechnieken onvoldoende mitosen kunnen worden geanalyseerd, zal men moeten overgaan tot in situ hybridisatie met specifieke probes (FISH techniek) om bepaalde numerieke chromosomale afwijkingen (zoals trisomie 8, monosomie 7, deletie 20q, verlies Y-chromosoom en afwijkingen chromosoom 17p (p53 locus) op te kunnen sporen.16,17 (Tabel 4)
Tabel 4. Frequent optredende chromosomale afwijkingen bij MDS
Ongebalanceerde afwijkingen Gebalanceerde afwijkingen
|
>10% |
5-8% |
3% |
1-2% |
1-3% |
18%^ |
|
+8* -7 of del(7q) -5 of del(5q) |
del(20q)* -Y* i(17q) of t(17p)
|
-13 of del(13q) del(11q) del(12p) of t(12p)
|
del (9q) idic(X)(q13) |
t(11;16) t(3;21) t(1;3) t(2;11) inv(3) t(6;9)
|
CN-LOH#17p |
* indien deze afwijkingen geïsoleerd voorkomen in afwezigheid van dysplasie worden deze niet als definitief bewijs voor MDS beschouwd; de niet geannoteerde afwijkingen kunnen bewijzend zijn voor MDS, ook als er geen dysplasie is.
# CN-LOH copy neutral loss of heterozygosity
^ in 18% van de patiënten met MDS met een complex afwijkend karyotype16,17
Immuunfenotypering
Immuunfenotypering van beenmerg voor de diagnose van MDS wordt aanbevolen. Zoals boven genoemd is het herkennen van dysplastische kenmerken door middel van cytomorfologie in één of meer hematopoïetische celreeksen niet altijd eenvoudig. In het bijzonder kunnen milde en discrete afwijkingen over het hoofd worden gezien. Flow cytometrische analyse van myeloïde voorlopers, erytroïde cellen en de myelo-monocytaire differentiatie in het beenmerg kan bijdragen aan het stellen van de diagnose MDS, Zie ook MDS Europe – Patient management (mds-europe.org).4,9,10,12 Bij het vinden van immuunfenotypische afwijkingen die passen bij MDS zonder morfologische dysplasie, zonder cytogenetische afwijkingen en bij afwezigheid van mutaties kan het reden zijn om beenmergonderzoek te herhalen bij blijvende verdenking op MDS. Er is gebleken dat in deze groep bij follow up vaker alsnog de diagnose MDS wordt gesteld.9,10 Patiënten die bij het eerste onderzoek geen morfologische aanwijzingen voor MDS hadden, maar wel met immuunfenotypering en/of genetisch onderzoek hadden 81% kans om twee jaar later alsnog gediagnosticeerd te worden met MDS.10Patiënten met een hoog risico ‘clonal hematopoiesis of undeterminate significance’ laten een vergelijkbare progressie vrije overleving zien als patiënten met een laag risico MDS.18 Dit zijn de patiënten die morfologisch onvoldoende dysplasie hebben voor de diagnose MDS, maar wel het klinisch beloop laten zien van een MDS. Verder onderzoek moet uitwijzen of afwijkingen gevonden met immuunfenotypering en moleculair onderzoek bij deze patiënten vervangend kan zijn voor morfologisch onderzoek voor de diagnose MDS.
Moleculair onderzoek
Moleculair onderzoek van het beenmerg bij het stellen van de diagnose MDS wordt aanbevolen. In de afgelopen jaren zijn een groot aantal DNA-mutaties geïdentificeerd die regelmatig gevonden worden bij MDS. Sommige mutaties worden bij +/- 10-30% van de patiënten gevonden (TET2, ASXL1, SF3B1, DNMT3A, SRSF2, RUNX1 en TP53), terwijl andere mutaties minder vaak voorkomen. De functie van de aangedane genen is divers, maar vaak betreft het genen die coderen voor eiwitten betrokken bij genexpressie zoals transcriptiefactoren en epigenetische regulatoren (TET2, DNMT3A, STAG2, EZH2, RUNX1 en ASXL1) of eiwitten die betrokken zijn bij RNA-splicing (SF3B1, SRSF2, U2AF1 en ZRSR2). De toegevoegde waarde van moleculair-genetische informatie voor het prognosticeren van MDS is onderzocht in een groot cohort van patiënten met MDS, waarbij de bekende prognostische parameters uit de IPSS-R werden gecombineerd met genmutatieanalyses. Hieruit is in 2022 de IPSS-M voortgekomen.3 Zie Module 3 Prognosticeren voor meer informatie over de IPSS-M. Naast implicaties voor de diagnostiek en het prognosticeren van MDS, kan moleculaire analyse ook behandelconsequenties hebben, bijvoorbeeld bij de aanwezigheid van een SF3B1 mutatie (zie module 4 behandeling van laag risico MDS).
Mutaties en klonale hematopoëse
Recent onderzoek heeft uitgewezen dat klonaal geëxpandeerde hematologische cellen die MDS/AML geassocieerde mutaties dragen frequent voorkomen bij personen die geen myeloïde maligniteit hebben. Dit fenomeen wordt klonale hematopoëse (clonal hematopoiesis, CH) genoemd. Het voorkomen en de grootte van deze kloons neemt toe met de leeftijd. Wanneer gemeten wordt met een gevoeligheid van 2% VAF (overeenkomend met een kloongrootte van 4% wanneer het een autosomaal gen betreft) wordt klonale hematopoëse aangetoond in 10-20 % van personen van 60 jaar. Rond de 80 jaar ligt dit op ongeveer 40%. Wanneer zeer gevoelige technieken worden gebruikt kunnen gemuteerde kloons bij het grootste deel van de gezonde populatie gemeten worden. Inmiddels wordt klonale hematopoëse met een VAF van 2% of hoger aangeduid met CHIP (clonal hematopoiesis of indeterminate potential). Personen met CHIP hebben een 5-10 maal hogere kans op het ontwikkelen van een myeloïde maligniteit binnen 5-10 jaar dan personen zonder CHIP. Bij dit verhoogde relatieve risico blijft het absolute risico evenwel bescheiden. Wanneer bij CHIP ook een cytopenie wordt gevonden wordt dit aangemerkt als CCUS en neemt het risico op een myeloïde maligniteit verder toe. Inmiddels zijn voor CCUS verschillende risicofactoren bekend (o.a. type gen mutatie, kloongrootte, aantal mutaties, trombocytengetal, MCV, RDW, leeftijd) die personen kunnen identificeren met een zeer sterk verhoogd risico op het ontwikkelen van een myeloïde maligniteit.18,19,20 Bij het diagnosticeren van een MDS kunnen gevallen van CCUS gevonden worden. Hoe het beste om te gaan met CCUS met een hoog risico op het ontwikkelen van een myeloïde maligniteit staat momenteel ter discussie.
Aanbeveling
Tabel 5
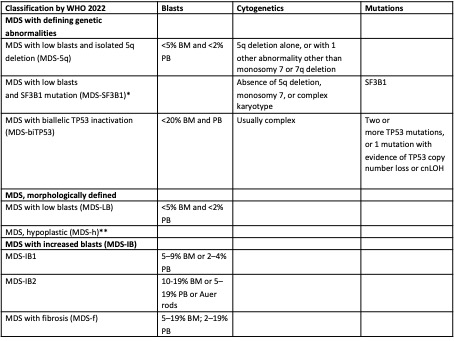
* Detection of ≥15% ring sideroblasts may substitute for SF3B1 mutation. Acceptable related terminology: MDS with low blasts and ring sideroblasts.
** By definition, ≤25% bone marrow cellularity, age adjusted.
BM bone marrow, PB peripheral blood, cnLOH copy neutral loss of heterozygosity.
Tabel 6
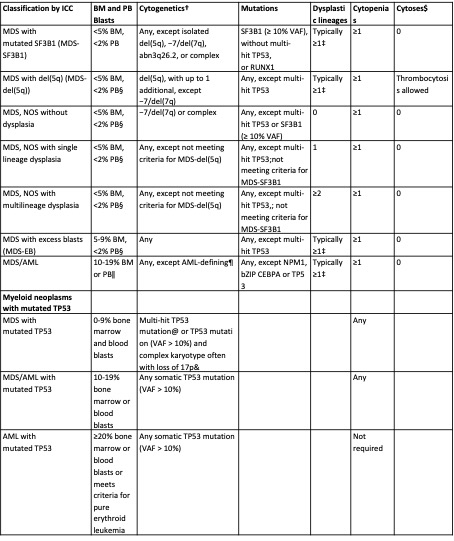
$ Cytoses: Sustained white blood count ≥ 13 × 109/L, monocytosis (≥0.5 × 109/L and ≥10% of leukocytes) or platelets ≥450 × 109/L; thrombocytosis is allowed in MDS-del(5q) or in any MDS case with inv(3) or t(3;3) cytogenetic abnormality.
† BCR::ABL1 rearrangement or any of the rearrangements associated with myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions exclude a diagnosis of MDS, even in the context of cytopenia.
‡ Although dysplasia is typically present in these entities, it is not required.
ǁ For pediatric patients (<18 y), the blast thresholds for MDS-EB are 5% to 19% in BM and 2% to 19% in PB, and the entity MDS/AML does not apply.
¶ AML-defining cytogenetics are listed in the AML section.
@Defined as 2 distinct TP53 mutations (each VAF > 10%) OR a single TP53 mutation with (1) 17p deletion on cytogenetics; (2) VAF of >50%; or (3) Copy-neutral LOH at the 17p TP53 locus.
& If TP53 locus LOH information is not available.
Onderbouwing
De werkgroep heeft ervoor gekozen om de meest recente WHO 2022 en ICC-classificatie op te nemen in de richtlijn omdat hierin de meest recente wetenschappelijk inzichten zijn verwerkt. De nadruk ligt op het onderscheiden van subgroepen van MDS op basis van cytogenetische en moleculaire afwijkingen indien aanwezig. Het werken met twee verschillende classificatiesystemen is omslachtig en het is belangrijk zo veel mogelijk ‘dezelfde taal te spreken’ in de behandeling van patiënten en wetenschappelijk onderzoek. Vanuit het ‘international consortium for myelodysplastic syndromes’ (icMDS) is een voorstel gedaan om beide classificaties te harmoniseren.21 Dit model geeft een versimpeling van de WHO 2022 en ICC en kan helpen in het classificeren van patiënten met MDS en is niet bedoeld ter vervanging van de WHO 2022 en/of ICC.
Aanbeveling
Het wordt aanbevolen de IPSS-M(olecular) te gebruiken voor het prognosticeren van en als leidraad voor therapiekeuze van patiënten met MDS. Voor het berekenen van de IPSS-M middels de IPSS-M Risk Calculator (mds-risk-model.com) zijn de volgende variabelen nodig: percentage blasten, hemoglobine, trombocyten, cytogenetica en moleculaire data. (SORT A)
Onderbouwing
Het beloop van MDS onderscheidt zich grofweg tussen beenmerg falen enerzijds en ontwikkelen van leukemie anderzijds. Het onderscheiden van deze twee groepen ligt aan de basis van het prognosticeren en voor de therapiekeuze van MDS.
In 2022 is de IPSS-M verschenen, de opvolger van de IPSS-R waarin mutaties een belangrijke rol spelen.3Mutaties in oncogenen kwamen in 90% van de patiënten voor in het IPSS-M cohort. Van de 152 genen die werden getest bleken 31 een toegevoegde waarde te hebben voor het bepalen van de prognose, bij een “variant allele frequency (VAF)” van 2% of groter. FLT3 (ITD en TKD) en MLL-PTD mutaties associeerden met een zeer slechte prognose, terwijl mutaties in SF3B1 correleerden met een relatief goede prognose. Van de 31 prognose-geassocieerde mutaties kregen er 18 een gen-specifieke score voor de risicoberekening, terwijl de 13 overige genen allen eenzelfde score in de IPSS-M kregen toebedeeld. Bijzonder was dat een enkelvoudige TP53 mutatie (TP53-mono) geen grote impact had op de prognose, terwijl meerdere TP53 mutaties bij dezelfde patiënt samengaat met een zeer slechte prognose. Het is daarom van belang om voor TP53 een assay te gebruiken die een TP53 mono- van een TP53 multi-hit kan onderscheiden. Wanneer niet alle 31 gen mutaties gemeten worden kan de IPSS-M score nog steeds worden berekend, maar de betrouwbaarheid van de risico inschatting neem dan af. Met de huidige gen panels die gebruikt worden kunnen doorgaans de meeste of alle 31 genen gemeten worden.
Het absolute neutrofielen aantal dat in de IPSS-R voorspellende waarde had, verliest die waarde wanneer de gen mutaties bekend zijn. Hetzelfde geldt voor geslacht.
Meerwaarde van de IPSS-M is dat-in tegenstelling tot de IPSS-R- de score ook toepasbaar is voor secundaire/therapie gerelateerde MDS. Ook is de IPSS-M toepasbaar bij patiënten die voorts behandeld worden met lenalidomide, hypomethylerende middelen of een stamceltransplantatie.
De werkgroep kiest ervoor om de IPSS-M aan te bevelen voor de prognosticeren van MDS, omdat deze in vergelijking tot de IPSS-R beter is in het voorspellen van de leukemie-vrije overleving, algehele overleving en risico op transformatie naar AML.3 De C-index van de IPSS-M is beter dan de C-index van de IPSS-R en daarmee geeft de IPSS-M een betere inschatting van het risico op leukemie en overlijden vergeleken met de IPSS-R.
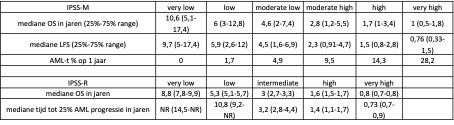
De IPSS-M onderscheidt 6 risicogroepen, in tegenstelling tot de IPSS-R die 5 risicogroepen onderscheidt. Zie tabel 7 voor de risicogroepen met bijbehorende overall survival en risico op progressie naar acute myeloïde leukemie.
In 46% van de patiënten leidde de her stratificatie volgens de IPSS-M ertoe dat patiënten van risicogroep wisselden. De meerderheid van deze patiënten (911/1223, 74%) werden ingedeeld in een hogere risicogroep. Belangrijk hierbij is dat in 7% van de gevallen patiënten twee of meer risico-categorieën opschuiven, hetgeen kan leiden tot andere behandelkeuze. Sinds het verschijnen van de IPSS-M is er retrospectieve data verschenen in hoeverre het gebruik van de IPSS-M zou leiden tot een andere therapiekeuze. In 17,4%-22,9% van de patiënten zou gebruik van de IPSS-M leiden tot intensiveren van de behandeling en in 4,8%-30,5% in niet intensieve behandeling. (Tabel 8)
De werkgroep beveelt aan om NGS toe te passen bij alle patiënten die jonger zijn dan 75 jaar en fit genoeg zijn voor een allogene stamceltransplantatie, om zo de mogelijkheid voor een potentieel curatieve behandeling niet te missen.
Verder is het zo dat lager risico patiënten met MDS en een SF3B1 mutatie, ongeacht de leeftijd in aanmerking komen voor behandeling met luspatercept indien erytropoëse stimulerende middelen (ESA) refractair of bij een lage vooraf kans op hematologische respons op ESA (zie bij behandeling) De aanwezigheid van een TP53 mutatie in combinatie met een complex karyotype is geassocieerd met een slechte prognose.22 Patiënten met een deletie 5q en een TP53 mutatie hebben een hoger risico op het ontwikkelen van een acute myeloïde leukemie en kortere overleving ten opzichte van patiënten met del5q zonder TP53 mutatie.23 Om die reden is het aan te bevelen om ten minste geïnformeerd te zijn over de mutatie status van SF3B1 en TP53. Het is in veel laboratoria mogelijk om materiaal in te laten vriezen en op een later moment alsnog NGS te laten inzetten.
Tabel 7
Tabel 8 Overzicht van studies naar therapeutische consequenties van het toepassen van de IPSS-M
|
|
n= |
% herstratificatie |
% upgrading |
% downgrading |
% intensificatie van behandeling |
% minder intensieve behandeling |
|
Bernard et al NEJM evidence 20223 |
2957 |
46 |
34 |
12 |
|
|
|
Zamanillo et al Frontiers in Oncology 202324 |
166 |
48 |
31 |
17 |
17 |
5 |
|
Lee et al Blood Cancer J 202325 |
649 |
43 |
29 |
13 |
15 |
2 |
|
Jáuregui et al Blood 202226 |
126 |
43 |
|
|
12 |
6 |
|
Sabile et al Leukemia&Lymphoma 202327 |
140 |
44 |
32 |
12 |
|
|
|
Baer et al Leukemia 202328 |
626 |
44 |
25 |
19 |
|
|
|
Sauta et al JCO 202329 |
2876 |
46 |
24 |
22 |
|
|
|
Huang et al Ann of Hematol 202430 |
798 |
45 |
29 |
16 |
|
|
|
Tentori et al JCO 202431 |
7118 |
|
|
19 |
15 |
Algemeen
De heterogeniteit van MDS maakt dat de behandeling een uitdaging is. De veelal oudere patiënt (gemiddelde leeftijd 74 jaar) met een MDS heeft naast zijn hematologische ziekte vaak andere aan leeftijd gerelateerde ziekten. De keuze van therapie wordt behalve door co-morbiditeit ook bepaald door andere individuele factoren zoals sociale situatie, vitaliteit en levensfilosofie van de patiënt. De richtlijnen voor behandeling zijn ingedeeld naar risicoprofiel gebaseerd op de huidige IPSS-M classificatie modellen. Onder “lager risico MDS” wordt verstaan: IPSS-M risicogroep “very low”, “low” of “moderate low” overeenkomende met IPSS-R risicogroep “very low” of “low”.31,32 Zie tabel 8 in module 3 met literatuur voor de onderbouwing en met name de publicatie van Tentori et al.31
In dit hoofdstuk wordt gebruik gemaakt van onderzoeksvragen met aanbevelingen per klinisch probleem (anemie, trombopenie, neutropenie) en de daarbij behorende onderbouwing. Bij meer dan één aanbeveling wordt gebruik gemaakt van een tabel voor een overzicht van de aanbevelingen met de SORT-gradering.
Behandeling voor patiënten met lager risico MDS
De behandeling van patiënten met lager risico MDS is primair gericht op het verbeteren van de bloedbeeldafwijkingen, met name anemie, en de daarmee samenhangende symptomen, om de kwaliteit van leven te verbeteren. Voor deze patiënten zijn, naast optimaal ondersteunende zorg, verschillende behandelopties beschikbaar die ingezet kunnen worden als er een behandelindicatie is. De verschillende behandelmodaliteiten worden hieronder besproken. Een klein deel van deze patiënten komt in aanmerking voor potentieel curatieve therapie in de vorm van een allogene stamceltransplantatie.
Welke patiënten met lager risico MDS komen in aanmerking voor allogene stamceltransplantatie?
Aanbeveling
Allogene stamceltransplantatie moet overwogen worden bij fitte patiënten met lager risico MDS indien sprake is van ernstige cytopenieën (ernstige persisterende neutropenie (arbitrair <0,3) of transfusie-afhankelijke anemie (≥ 2 units / maand, gedurende 6 maanden) en/of trombopenie < 30 x 10 ^9/L) zonder respons op andere therapieën (bijvoorbeeld ESA), dan wel in aanwezigheid van prognostisch ongunstige factoren zoals ongunstige cytogenetica en/of gen mutaties (dit betekent in het algemeen een hoge IPSS-M score), ernstige beenmergfibrose of erfelijke predispositie voor myeloïde maligniteiten. (SORT C)
Onderbouwing
Een allogene stamceltransplantatie is de enige curatieve behandeloptie voor patiënten met MDS. Zie hiervoor ook de behandeling van hoger risico MDS. Gezien de therapie-gerelateerde morbiditeit en mortaliteit is een goede selectie van patiënten die hiervoor in aanmerking komen essentieel. Het aantal studies dat relevant is voor het beantwoorden van de vraag is zeer beperkt. Het aantal geïncludeerde patiënten in onderzoek is klein. De publicaties rapporteren met name retrospectieve analyses. De gebruikte bronnen zijn expert opinion, de ESMO guidelines een recent gepubliceerde retrospectieve studie en de website van MDS Europe (https://mds-europe.org).31,33-35
De groep patiënten met lager risico MDS is heterogeen. Bij de afweging om een transplantatietraject in te gaan spelen enerzijds patiënt-gerelateerde factoren zoals performance status en co-morbiditeiten een rol en anderzijds ziekte-gerelateerde kenmerken naast donor-beschikbaarheid en -kenmerken. Bij patiënten met ernstige neutropenie en/of transfusie-afhankelijke anemie of trombopenie moeten het risico en de belasting van de cytopenieën en transfusies worden afgewogen tegen het risico van een allogene SCT. Hierbij spelen de mate van neutropenie (in de richtlijn van MDS Europe wordt een neutrofielen aantal <0,3 x 10*/L als ernstig beschouwd) en de transfusiebehoefte een rol evenals de reactie op medicamenteuze therapie (zoals ESA). De snelheid waarmee cytopenieën zich ontwikkelen dan wel toenemen heeft eveneens prognostische waarde. Verschillende cytogenetische afwijkingen en gen mutaties zijn geassocieerd met een slechtere prognose bij MDS, ook in de lager risico IPSS-R groepen. Om die reden is het belangrijk bij lager risico MDS patiënten die potentieel in aanmerking komen voor transplantatie de IPSS-M score te bepalen en mee te wegen bij de beslissing. Een recent ontwikkeld beslissingsmodel voor SCT bij MDS liet zien dat, gebruik makend van de IPSS-M score, bij 17% van de patiënten het beleid ten aanzien van SCT (gebaseerd op de IPSS-R score) veranderde, resulterende in een overlevingsvoordeel.31
Bij een deel van de MDS patiënten is sprake van fibrotische veranderingen in het beenmerg. De WHO 2022 beschouwt MDS met fibrose (MDS-f, geduid als reticuline vervezeling graad ≥ 2) als een aparte ziekte entiteit.5,36 Ernstige fibrose heeft een negatieve impact op de overleving van deze patiënten, ook na een allogene stamceltransplantatie.37 Om die reden wordt geadviseerd om de mate van fibrose (en zo mogelijk snelheid van progressie van fibrose) mee te nemen in de overweging om over te gaan tot een allogene stamceltransplantatie.
Bij patiënten bij wie sprake is van een erfelijke predispositie voor myeloïde maligniteiten heeft ook bij een vroeg stadium van MDS een allogene stamceltransplantatie soms de voorkeur boven een ondersteunend beleid. Dit wordt voor de verschillende vormen van erfelijke predispositie niet verder besproken in deze richtlijn. Het is belangrijk patiënten met (verdenking op) een erfelijke vorm van MDS te verwijzen naar een centrum voor tertiaire zorg.
Welke behandelmogelijkheden zijn er voor anemie?
Aanbevelingen
|
SORT Grade |
Conclusie* |
|
B |
Er is geen algemene Hb-trigger voor erytrocytentransfusies bij MDS |
|
A |
Erytropoëse-stimulerende middelen (ESA) leiden tot verbetering van de anemie in 20 tot 74% van MDS patiënten, afhankelijk van de transfusiebehoefte en endogene erytropoëtine spiegel |
|
A |
Lenalidomide bij patiënten met een geïsoleerde del(5q) leidt bij 50-70% tot transfusie-onafhankelijkheid |
|
A |
Lenalidomide bij non del(5q) leidt tot een respons bij ongeveer 20-30% van de patiënten |
|
B |
Behandeling met ATG/ciclosporine leidt bij een kleine subgroep van patiënten met hypoplastische MDS tot een verbetering van cytopenieën. |
|
A |
Luspatercept leidt tot transfusie-onafhankelijkheid bij 38% van de patiënten met MDS met SF3B1 mutatie (of MDS met laag blasten aantal en ≥15% ringsideroblasten) |
|
B |
Behandeling met azacitidine is niet geregistreerd voor patiënten met lager risico MDS. Het kan in uitzonderingsgevallen overwogen worden, met name bij ernstige inflammatoire klachten. |
|
B |
Imetelstat kan leiden tot transfusie onafhankelijkheid in 40% van de patiënten met (non del5q) MDS. |
*conclusie = antwoord op de onderzoeksvraag
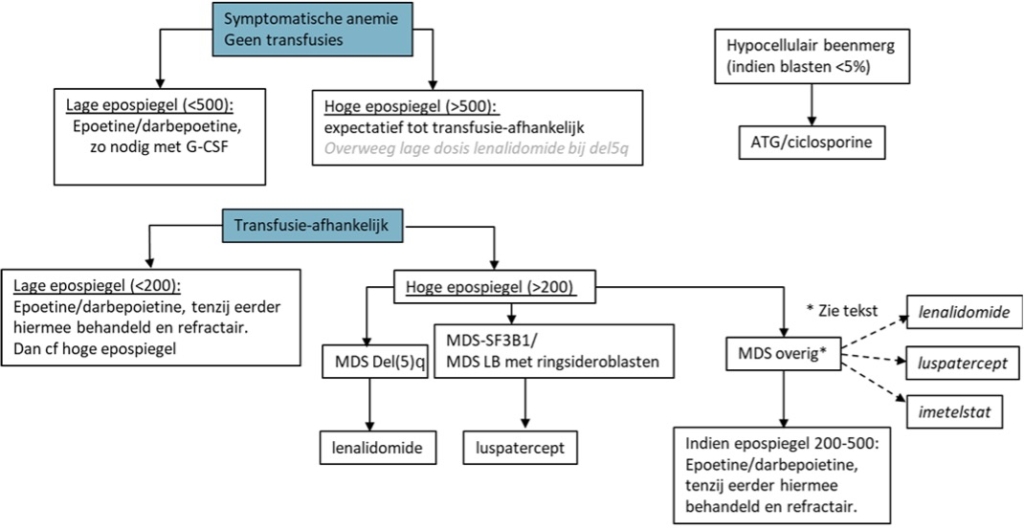
Figuur 2 Schematisch overzicht van de behandelmogelijkheden van anemie bij patiënten met laag risico MDS. Voor lenalidomide is de add-on aanvraag ingediend bij verschijnen van deze richtlijn. Imetelstat is EMA goedgekeurd, maar er is nog geen vergoeding.
Onderbouwing
Symptomatische anemie is het meest voorkomende symptoom bij lager risico MDS patiënten en kan grote impact op de kwaliteit van leven hebben.
Rodebloedceltransfusie
Rode bloedcel transfusie is de meest gebruikte methode ter verhoging van het Hb-gehalte en wordt het meest toegepast bij symptomatische anemie bij MDS. De juiste Hb-trigger voor transfusie is niet duidelijk gedefinieerd. Dit geldt eveneens voor transfusiefrequentie en aantal eenheden per keer. Het transfusieschema dient individueel te worden bepaald met inachtneming van co-morbiditeit van de veelal oudere patiënt met MDS en de gepercipieerde kwaliteit van leven. Een Hb onder de 4.4 mmol/l is eigenlijk altijd symptomatisch. Langdurige ernstige anemie is geassocieerd met cardiaal falen en verminderde kwaliteit van leven. Naast directe bijwerkingen van een transfusie zoals een transfusiereactie en volume overbelasting, is er op langere termijn een risico op allo immunisatie en ijzerstapeling. Er zijn geen aanwijzingen dat rode bloedceltransfusies ziekteprogressie negatief beïnvloedt.
Voor specifieke adviezen over selectie van bloedproducten wordt verwezen naar de CBO-richtlijn Bloedtransfusiebeleid.
Erytropoëtine (ESA) en granulocyten-stimulerende factoren (G-CSF)
Bij een symptomatische anemie dient, indien mogelijk al voor start van bloedtransfusies, overwogen te worden de patiënt te behandelen met ESA eventueel in combinatie met G-CSF. Het streef Hb bij starten van ESA met/zonder G-CSF is het bereiken van een Hb 6.2-7.4 mmol/L.38 De beste respons van behandeling met ESA en/of G-CSF treedt op bij patiënten met lager risico MDS die nog niet of beperkt transfusie behoeftig zijn (< 2 RBC/maand) en een lage endogene erytropoëtine spiegel (<500 U/l) hebben. De geschatte kans op respons bij met Epo/G-CSF behandelde patiënten met een transfusiebehoefte < 2 RBC/maand en een Epo < 500 U/l is ongeveer 74%.39-41 Patiënten met of een Epo > 500 U/l of een transfusiebehoefte van >2 RBC/maand hadden een lagere respons kans (23%).39 Hoewel een ondergrens van Epo van 500 U/l gehanteerd wordt, aangezien deze drempel als enige gevalideerd is in een predictie model voor respons op Epo/G-CSF, laten studies zien dat een drempel van de endogene erytropoëtine spiegel van 100 U/l en (zeer) laag risico MDS een betere kans op respons op Epo +/- G-CSF voorspelt.42,43 In het bijzonder bij het subtype met ringsideroblasten is aangetoond dat toevoeging van G-CSF bijdragend is.44 De mediane responsduur van de behandeling met Epo +/- G-CSF is ongeveer 24 maanden.45 De meest gebruikte Epo’s zijn erytropoëtine-α, erytropoëtine-β en het langwerkende darbepoëtine. 30.000 IE van erytropoëtine-α en erytropoëtine-β komt overeen met 150 μg darbepoëtine. De startdosering is 30.000 IE s.c. 1x per week of equivalente dosering 150 μg darbepoëtine/week [of 300 μg/2 weken of 500 μg/3 weken]. Indien na 8 weken het Hb gehalte niet stijgt met tenminste 0.6 mmol/l, wordt de dosering verhoogd tot 60.000 IE erytropoëtine-α/erytropoëtine-β of 300 μg darbepoëtine. Als na nog eens 8 weken geen stijging in het Hb gehalte is bereikt, kan gestart worden met G-CSF (filgrastim); 300 μg 1-3 x week op geleide van het leukocyten aantal. Indien het leukocyten aantal boven de 30×109/l stijgt, wordt kortdurend gestopt en kan G-CSF worden herstart met een dosis reductie van 50% na normalisering van het leukocyten aantal. Indien na 6 weken geen stijging in het Hb gehalte is vastgesteld, is verdere behandeling met Epo/G-CSF niet zinvol. De behandeling met groeifactoren leidt bij responderende patiënten tot een overlevingsvoordeel en verbetering van kwaliteit van leven. Een versnelde progressie van MDS naar AML, als gevolg van gebruik van deze groeifactoren, is in diverse onafhankelijke studies niet aangetoond. De behandeling met ESA mag geen vertraging veroorzaken in de keuze voor een allogene stamceltransplantatie voor patiënten die hiervoor in aanmerking komen.
Lenalidomide bij MDS del(5q)
Voor MDS patiënten met (geïsoleerde) del(5q) is lenalidomide een effectief geneesmiddel in een dosering van 10 mg per dag en is hiervoor als zodanig geregistreerd, mits transfusie-afhankelijk, refractair op ESA en/of G-CSF, en/óf endogene epo spiegel > 200 U/L. Effectieve schema’s zijn lenalidomide 10 mg/dag op dag 1-21 (elke 4 weken) of 5 mg/dag op dag 1-28 (elke 4 weken). Eventueel kan de dosis van 10 mg worden verlaagd naar 5 mg als er een respons (transfusie-onafhankelijkheid of stijging van het Hb) is opgetreden. Het bloedbeeld dient de eerste 8-12 weken regelmatig en daarna minimaal maandelijks gecontroleerd te worden. In een belangrijke studie die in 2006 is gepubliceerd werd 67% van de patiënten onafhankelijk van transfusie en kreeg zelfs 44% van de patiënten een complete cytogenetische respons. In deze studie was de mediane duur tot respons 4,6 weken. Deze responsen houden vaak jaren aan.46 In een gerandomiseerde fase-III studie (MDS-004) bij transfusie afhankelijke patiënten met een MDS en del(5q) chromosomale afwijking, waarbij gerandomiseerd werd voor lenalidomide (in 2 doseringen versus placebo) konden deze resultaten worden bevestigd.47 In een Nederlands ‘named patient program’ hebben we laten zien dat patiënten met MDS met del(5q) die een hematologische respons laten zien (HI-E) en CR bereiken, een verbeterde overleving hebben ten opzichte van hen die geen CR bereiken.48 In de WHO2016 classificatie wordt er een categorie gedefinieerd met een chromosoom 5 afwijking waarbij naast de del(5q) tevens sprake mag zijn van 1 additionele afwijking tenzij deze afwijking een monosomie 7 of del7q betreft. Dit is gebaseerd op het feit dat de prognose en respons op therapie met lenalidomide bij deze patiënten gelijk is aan die van patiënten met MDS met een geïsoleerde del(5q). Mutaties in TP53 zijn geassocieerd met ziekteprogressie onder behandeling met lenalidomide. Daarnaast is het van belang dat bij patiënten die lenalidomide resistentie hebben ontwikkeld, bij 20% een TP53 mutatie wordt aangetoond.49,50 De werkgroep adviseert patiënten met een TP53 mutatie dan ook niet met lenalidomide te behandelen. Indien toxiciteit op lenalidomide wordt vermoed zich uitend in toenemende cytopenie, wordt aanbevolen ziekteprogressie als oorzaak uit te sluiten. Indien patiënten hun respons op lenalidomide verliezen en er geen aanwijzingen zijn voor ziekteprogressie, zoals bijvoorbeeld het ontstaan van een TP53 mutatie, kan na een ‘drug holiday’ overwogen worden om opnieuw behandeling met lenalidomide te proberen.51
Daarnaast is in een fase 3 gerandomiseerde trial (SintraREV) aangetoond dat het starten van lenalidomide bij MDS met del(5q) voor het optreden van transfusieafhankelijkheid vs placebo, de tijd tot transfusiebehoefte verbeterd (mediane tijd niet behaald vs 11.6 maanden resp). Hiervoor bestaat echter geen registratie.52
Lenalidomide bij MDS non-del(5q)
De prospectieve, fase 3 MDS-005 studie in MDS-non-del(5q) waarbij werd gerandomiseerd tussen lenalidomide en placebo liet op 24 weken een transfusie onafhankelijkheid zien van 17,5% voor de lenalidomide arm met een mediane responsduur van 7,2 maanden voor patiënten die ≥ 8 weken respondeerden.54 Een prospectieve fase III studie van het Franse MDS consortium (GFM) toonde vergelijkbare resultaten in non-del(5q) MDS waarbij het upfront toevoegen van ESA een toename in transfusie onafhankelijkheid liet zien (24% vs 13.8%) zonder dat dit effect had op EFS en OS.53
In de HOVON89 studie werden 30 (16%) patiënten met del(5q) en 154 (84%) patiënten met non-del(5q) geïncludeerd en gerandomiseerd tussen behandeling met lenalidomide +/- ESA/G-CSF.54 Bij 73% van de del(5q) en 30% van de non-del(5q) patiënten werd transfusie-onafhankelijkheid bereikt, na mediaan 2.8 en 3.2 maanden respectievelijk. Er werd geen voordeel aangetoond voor toevoeging van ESA/G-GSF, alhoewel bij deze studie ESA/G-CSF pas werd toegevoegd na 4 kuren lenalidomide indien onvoldoende respons bereikt werd.
Een add-on aanvraag voor de vergoeding van lenalidomide bij MDS met non-del(5q) is ingediend.
Immuunsuppressieve therapie
Immuunsuppressieve therapie met paarden anti-thymocytenglobuline (ATGAM) en ciclosporine lijkt voornamelijk nuttig bij patiënten met een lager risico MDS en een hypocellulair beenmerg, zoals bleek uit een internationaal cohort van 207 MDS patiënten.55 Hierbij werd een respons gezien in bijna 50% van de patiënten, en bij 30% werd transfusie onafhankelijkheid bereikt. Andere mogelijke voorspellers voor respons (zoals aanwezigheid van HLA-DR15 genotype of de aanwezigheid van een (kleine) PNH kloon zijn) zijn controversieel.55,56 Deze behandelingen leiden tot een respons van meerdere cellijnen en kunnen daarom overwogen worden bij patiënten met een symptomatische anemie, neutropenie en/of trombopenie. Responsen kunnen tot wel 6 maanden na start van de ATG/ciclosporine optreden en kunnen langdurig aanhouden. ATG dient gegeven te worden in centra met ervaring in het gebruik hiervan. Daarnaast werd in een retrospectieve analyse (n= 16) in MDS patiënten met een beeld van beenmergfalen met een geïsoleerde del(13q) of del(13q) met een andere genetische afwijking, bij 100% en 40% respectievelijk een respons gezien op immuunsuppressie (ATG en/of ciclosporine), met een mediane 10 jaars overleving van 83% en 67% resp. (Hosokawa et al Haematologica 2012)
Luspatercept
Luspatercept is zowel door FDA (2020) als EMA (2021) goedgekeurd voor de behandeling van transfusie-afhankelijke patiënten met MDS met een SF3B1 mutatie en/of ≥ 15% ringsideroblasten (RS), die reeds ESA-refractair zijn. Luspatercept stimuleert de erytropoëse via blokkade van de SMAD2/3 signaaltransductieroute. Deze signaaltransductieroute heeft een remmend effect op de uitrijping van erytroïde voorlopercellen. Een gerandomiseerde, placebogecontroleerde studie met luspatercept bij zeer laag tot intermediair risico MDS met ringsideroblasten (ESA refractair dan wel lage responskans op ESA) liet zien dat 38% van de patiënten minimaal 8 weken transfusie-onafhankelijk werd en 33% meer dan 12 weken. De responskans was het grootst bij de groep met de laagste transfusie-afhankelijkheid (<4 Eh per 8 weken). Luspatercept wordt eenmaal per 3 weken subcutaan toegediend. Meest voorkomende bijwerkingen zijn vermoeidheid, asthenie, diarree, misselijkheid en duizeligheid.57 Daarnaast is luspatercept ook vergeleken met ESA in een fase 3 studie in de eerste lijn (COMMANDS trial) waarin laag risico MDS patiënten deelnamen met én zonder ringsideroblasten.58Patiënten met del(5q) kwamen niet in aanmerking voor deelname. Transfusieonafhankelijkheid werd gezien bij 58% van de luspatercept groep versus 31% van de ESA-groep. In een subgroep analyse bij patiënten zonder ringsideroblasten bleek luspatercept niet vaker tot transfusie onafhankelijkheid te leiden vergeleken met ESA (41% vs 46%).59 Daarom wordt het gebruik van luspatercept niet geadviseerd bij deze groep in de eerste lijn. Er zijn nog onvoldoende gegevens over luspatercept in de tweede lijn bij MDS patiënten zonder ringsideroblasten om hierover aanbevelingen te kunnen doen.
Azacitidine
Azacitidine is in Europa alleen geregistreerd voor de behandeling van IPSS-intermediair 2 en hoog risico MDS patiënten. Een meta-analyse van studies naar het effect van azacitidine bij transfusie-afhankelijke lager risico MDS patiënten laat een grote variatie zien tussen de verschillende studies (zowel wat betreft geïncludeerde patiënten, methodologie als resultaten).60 Verreweg de meeste patiënten in de meta-analyse waren afkomstig van de AVIDA studie.61 Het in de meta-analyse geschatte percentage patiënten dat transfusie-onafhankelijk wordt is 38%. Geen eerdere behandeling met ESA was een voorspellende factor voor respons. Met azacitidine kunnen ook bij lager risico MDS patiënten positieve effecten op het trombocytenaantal bereikt worden. Meest voorkomende bijwerking is echter verergering van cytopenie(ën). Daarnaast zijn auto-inflammatoire aandoeningen, zoals artritis, vasculitis en Sweet syndroom geassocieerd met MDS of toe te schrijven aan het VEXAS syndroom. Vanwege het immuun modulerende effect van azacitidine op onder andere de NK-cellen, T-cellen en dendritische cellen, is eerder onderzoek gedaan naar het effect hiervan op deze auto-inflammatoire klachten. Een kleine retrospectieve studie liet bij 86 % afname zien van auto-inflammatoire klachten.62 Een prospectieve studie van GFM is recent uitgevoerd ter beoordeling van dit effect. Resultaten hiervan worden nog verwacht (NCT 02985190).
Een overlevingsvoordeel van behandeling met azacitidine bij laag-risico MDS is niet bewezen. Er is onvoldoende bewijs en geen vergoeding om behandeling met azacitidine standaard aan te bevelen bij lager risico MDS patiënten. Het kan in uitzonderingsgevallen overwogen worden bij ernstige inflammatoire klachten.
Imetelstat
Imetelstat is een oligonucleotide, dat via RNA-binding de activiteit van telomerase remt. Cellen met hoge telomerase activiteit zijn gevoelig voor de effecten hiervan. Effectiviteit en veiligheid zijn onderzocht in een fase 3 dubbel-blinde, placebogecontroleerde studie. Patiënten met een IPSS laag of intermediair-1 risico MDS (geen del(5q)) werden geïncludeerd indien ze transfusie-afhankelijk waren (>4 eenheden/8 weken) en refractair op ESA/endogene epospiegel >500.63 Belangrijke exclusiecriteria waren een neutrofielenaantal <1.5 x 10*9/L en trombocyten <75 x 10*9/L. Imetelstat wordt eenmaal per 4 weken intraveneus toegediend. Het primaire eindpunt van transfusie-onafhankelijkheid gedurende tenminste 8 weken werd bereikt in 40% (47/118) van de patiënten in de imetelstat-groep versus 15% (9/60) in de placebogroep. Na 24 weken betrof dit percentage 28% versus 3% en na 1 jaar 18% versus 2%. Belangrijkste bijwerkingen waren neutropenie en trombopenie (tot graad 3-4), leverenzymstoornissen en hoofdpijn. Imetelstat is door de EMA goedgekeurd voor lager risico MDS en een transfusie-afhankelijk anemie. In Nederland is imetelstat op dit moment niet beschikbaar. De werkgroep ziet een rol voor imetelstat in de toekomst bij patiënten met (niet del5q) MDS. Dit betreft de volgende categorie patiënten: 1. Bij transfusie-afhankelijk patiënten met SF3B1/ringsideroblasten in de derde lijn bij onvoldoende (kans op) respons op ESA en luspatercept 2. Bij de overige MDS patiënten indien onvoldoende (kans op) respons op ESA, én een contra-indicatie voor/ineffectiviteit van lenalidomide (indien in de toekomst vergoed) én geen belangrijke contra-indicaties voor imetelstat, zoals diepe granulopenie of trombopenie.
Welke behandelmogelijkheden zijn er voor trombopenie?
Aanbevelingen
|
SORT Grade |
Conclusie* |
|
C |
Er is geen bewijs dat profylactische trombocytentransfusies bij lager risico MDS patiënten die ondersteunende zorg ontvangen leiden tot minder bloedingscomplicaties |
|
B |
Trombopoëse-stimulerende middelen kunnen een positief effect hebben op MDS-gerelateerde trombopenie. |
|
C |
Het is onvoldoende duidelijk of trombopoëse-stimulerende middelen kunnen leiden tot ziekteprogressie bij MDS |
|
B |
Behandeling met ATG/ciclosporine leidt bij een kleine subgroep van de MDS patiënten tot een verbetering van cytopenieën. |
|
B |
Azacitidine kan bij een subgroep van lager risico MDS patiënten leiden tot verbetering van de cytopenieën. Toename van cytopenieën is de meest voorkomende bijwerking. |
*conclusie = antwoord op de onderzoeksvraag
Onderbouwing
Trombopenie treedt op bij ongeveer 30% van de lager risico MDS patiënten.
Trombocytentransfusie
Trombocytentransfusies bij trombopenie zijn alleen geïndiceerd tijdens acute bloedingsepisoden of profylactisch bij diagnostische invasieve of chirurgische ingrepen. Het standaard toedienen van trombocytenconcentraten bij patiënten met een trombopenie (<10×109/l) zonder bloedingen kan leiden tot antistofvorming tegen plaatjes en is daarom niet geïndiceerd. Voor specifieke adviezen over selectie van bloedproducten, en adviezen bij gebruik bloedverdunners, wordt verwezen naar de richtlijn Bloedtransfusiebeleid.
Trombopoëse-stimulerende middelen
In een gerandomiseerde, placebogecontroleerde fase twee studie naar het effect van romiplostim werd 48% van de patiënten in de romiplostim-arm onafhankelijk van trombocytentransfusies.64,65 Bloedingscomplicaties waren minder frequent in de responderende patiënten. Vanwege aanwijzingen dat behandeling zou kunnen leiden tot ziekteprogressie (4/44 patiënten liet een tijdelijke toename van blasten in het beenmerg zien, 2 patiënten ontwikkelden AML tijdens de studie) is de behandeling met romiplostim voortijdig stopgezet. Het is niet duidelijk of er een oorzakelijk verband is tussen romiplostim en de ziekteprogressie. Een fase 2 studie met eltrombopag waarbij 169 patiënten werden geïncludeerd en gerandomiseerd tussen eltrombopag en placebo toonde ook een positief effect op het trombocytengetal en de incidentie van (WHO graad ≥ 2 bloedingsscore) bloedingscomplicaties.66 Alle patiënten hadden bij aanvang trombocyten < 30 x 10 ^9/L en 30% was trombocyten- transfusie afhankelijk. Transfusie onafhankelijkheid werd bereikt bij 54% en een trombocyten respons werd gezien bij 42%. De incidentie van ontwikkeling van AML was in deze studie niet groter in de eltrombopag groep versus de placebogroep. Zowel romiplostim als eltrombopag zijn niet geregistreerd voor de behandeling van MDS-gerelateerde trombopenie.
Deze middelen kunnen worden overwogen bij patiënten indien er een duidelijke auto-immuun trombopenie aanwezig is in het kader van een MDS-geassocieerde ITP, die refractair is op prednison. De werkgroep is van mening dat eltrombopag van meerwaarde kan zijn bij lager risico MDS patiënten, zonder toename van blasten en zonder fibrose, met een persisterende diepe trombopenie (<30 x 10*9/L). Aan het voorschrijven van eltrombopag zijn geen voorwaarden verbonden en is de vergoeding volgens het geneesmiddelen vergoedingssysteem.
Immuunsuppressieve therapie
Zie Immuunsuppresieve therapie bij anemie.
Azacitidine
Zie Azacitidine bij anemie.
Welke behandelmogelijkheden zijn er voor granulopenie?
Aanbevelingen
|
SORT Grade |
Conclusie* |
|
C |
Er is geen bewijs dat behandeling met G-CSF de levensduur van MDS patiënten verlengt. |
|
B |
Behandeling met ATG/ciclosporine leidt bij een kleine subgroep van de MDS patiënten tot een verbetering van cytopenieën. |
*conclusie = antwoord op de onderzoeksvraag
Onderbouwing
Een diepe neutropenie komt voor bij een minderheid van de lager risico MDS patiënten.
G-CSF
Bij patiënten met een neutropenie kan G-CSF worden overwogen ter voorkoming van recidiverende infecties, hoewel data die het effect hiervan beoordelen schaars zijn. De dosering G-CSF zal worden getitreerd van 300 μg 1x/dag tot 1-3 x week op basis van het absolute aantal neutrofielen (ANC); Het doel van deze behandeling is niet het normaliseren van het aantal neutrofielen, maar het streven naar een ANC van >0,5×109/l én het voorkomen van (ernstige) infecties. Ondanks het feit dat er geen data ter beschikking staan lijkt het gebruik van het gepegyleerde G-CSF (Pegfilgrastim) welke bij de chemotherapie geïnduceerde neutropenie van belang is, minder rationeel bij patiënten met MDS. De farmacologische beschikbaarheid van pegfilgrastim is namelijk (omgekeerd) gerelateerd aan de aanwezigheid van neutrofiele granulocyten.
Immuunsuppressieve therapie
Zie Immuunsuppressieve therapie bij anemie
Azacitidine
Zie azacitidine bij anemie
Wanneer moet ijzerchelatie worden gestart?
Aanbeveling
IJzerchelatie moet overwogen worden bij een ferritine >1000 ug/l als de levensverwachting 3 jaar of langer is volgens de IPSS-M en er geen contra-indicaties voor chelatietherapie zijn.
|
SORT Grade |
Conclusie* |
|
C |
IJzerstapeling kan leiden tot orgaanschade en een verminderde overleving op langere termijn. |
|
C |
IJzerchelatie lijkt de overleving van MDS patiënten met secundaire ijzerstapeling te verbeteren |
*conclusie = antwoord op de onderzoeksvraag
Onderbouwing
Bij elke rode bloedceltransfusie neemt de ijzervoorraad toe met ongeveer 200 mg. Lager risico MDS patiënten krijgen vaak gedurende meerdere jaren bloedtransfusies toegediend en lopen daarom het risico op secundaire hemochromatose. Bij patiënten met een MDS met ringsideroblasten is bij diagnose de ijzervoorraad vaak al verhoogd ten gevolge van een gestoorde ijzerinbouw.
IJzerchelatie
Progressieve ijzerstapeling bij MDS wordt niet alleen veroorzaakt door rode bloedceltransfusies. Een groot gedeelte van deze patiënten hebben vanwege ineffectieve erytropoëse, analoog aan congenitale hemoglobinopathie, ook ijzerstapeling zonder rode bloedcel transfusies. Dit is met name het geval bij MDS met ringsideroblasten en/of een SF3B1 mutatie. Bij MDS zijn er aanwijzingen dat ijzerstapeling ook daadwerkelijk klinisch relevante orgaanschade geeft.67 In ieder geval is er een negatieve relatie tussen de hoogte van het serum ferritine gehalte en overleving.68,69 Een verhoogd serum ferritine en labiel plasma ijzer (LBI) is eveneens gecorreleerd met een slechtere uitkomst na een allogene stamceltransplantatie. Bij patiënten met een ferritinegehalte boven de 1000 ug/l is het advies ijzerchelatie te geven mits de levensverwachting van de MDS patiënten op basis van de IPSS tenminste 3 jaren betreft. Naast het ferritinegehalte kunnen ook de hoogte van de transferrinesaturatie (boven of onder de 80%) en de mate van ijzerstapeling (LIC; liver iron content) op een MRI-lever of MRI-hart worden meegenomen in de overwegingen om ijzerchelatie te starten.70 Interessant is de observatie dat bij een deel van de patiënten met MDS die ijzerchelatietherapie ontvangen, een verbetering van de hematopoïese wordt waargenomen, die zelfs tot transfusieonafhankelijkheid kan leiden. De enige prospectieve gerandomiseerde studie (TELESTO) welke de rol van ijzerchelatie door middel van deferasirox bij het laag en intermediair-1 risico MDS onderzoekt, laat een significante reductie in het aantal ijzergerelateerde events voor de deferasirox arm zien versus de placebo-arm.71 Kanttekening bij deze studie is dat de studie veel minder mensen includeerde dan oorspronkelijk beoogd en werd aangepast van een fase 3 naar fase 2 studie. Het Europese MDS consortium heeft in een prospectieve registratie studie aangetoond dat ijzerchelatie een gunstig effect laat zien op PFS en OS.72 Er is onvoldoende bewijs voor testen om het effect van ijzerchelatie te meten. Serum ferritine wordt het meest gebruikt voor ijzerchelatie monitoring. De grenswaarde is echter niet gedefinieerd. Het laten dalen en stabiliseren van het ferritine tot <1000 ug/L lijkt een goede richtlijn. Ook verbetering van de LIC op een MRI-lever en/of hart kan hierin meegenomen worden. In de afweging om met ijzerchelatie te starten moeten de mogelijke bijwerkingen van ijzerchelatie, zoals het optreden van nierfunctievermindering, worden meegenomen.