Terug naar het richtlijnenoverzicht
Primaire myelofibrose (PMF) behoort samen met essentiële trombocytemie (ET) en polycythemia vera (PV) tot de Philadelphia chromosoom-negatieve myeloproliferatieve aandoeningen (MPN). Het is een relatief zeldzame aandoening met een geschatte incidentie van 0,5-1,5 per 100.000 inwoners per jaar. De ziekte manifesteert zich veelal op middelbare leeftijd in gelijke mate bij mannen en vrouwen. PMF kan primair zijn, maar zich ook secundair ontwikkelen na ET of PV (post-PV MF, post-ET MF). Bij 50-60% van de PMF patiënten kan een JAK2V617F mutatie aangetoond worden, bij ongeveer 30% een CALR mutatie en bij 8% een MPL mutatie. Ongeveer 12% van de patiënten heeft geen van deze 3 mutaties (triple negatief). De ziekte begint met een initiële prefibrotische/vroege fase (pre-PMF), waarbij er hypercellulair beenmerg is zonder of met minimale fibrose en ontwikkelt zich in de tijd tot een fibrotisch stadium (overte PMF) met kenmerkend leuco-erytroblastair bloedbeeld met traandruppel cellen, hepatomegalie en splenomegalie.Swerdlow 2017 De bestaande richtlijn dateert uit 2018 en was toe aan actualisatie vanwege herziening van de diagnostische criteria in 2016 en herziening van de richtlijnen van European Leukemia Net (ELN).Arber 2016, Barbui 2018 De richtlijn is grotendeels gebaseerd op deze aanbevelingen. De huidige herziening is vanwege nieuwe WHO criteria, gewijzigde vergoeding van lenalidomide en tekstuele afstemming met de andere MPN richtlijnen.Khoury, 2022
Deze richtlijn is bedoeld ter optimalisering van de diagnostiek, behandeling en poliklinisch vervolg van PMF toepasbaar in alle Nederlandse ziekenhuizen (geen echelonering van toepassing).
Deze richtlijn is bestemd voor alle professionals die betrokken zijn bij de diagnostiek, behandeling en begeleiding van patiënten met PMF, zoals internist-hematologen, internist-oncologen, internisten, etc.
Totstandkoming
Voor het ontwikkelen van de richtlijn is in 2017 de richtlijnwerkgroep MPN van de CML/MPN werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met MPN. Het betreft hierbij een aanpassing/actualisatie van de reeds bestaande richtlijn uit 2018.
Op voorstel van de richtlijnbeheerder dr. P.A.W. te Boekhorst heeft de richtlijnwerkgroep de conceptrichtlijnen per e-mail en fysieke bijeenkomsten becommentarieerd en aangepast. De uitgangsvragen werden hierbij besproken en aanbevelingen gemaakt.
De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Procedure voor commentaar en autorisatie
Een conceptversie van de richtlijn werd op 04-09-2023 voor commentaar aangeboden aan de leden van de Nederlandse Vereniging voor Hematologie (NVvH). Commentaar geeft input vanuit het veld om de kwaliteit en de toepasbaarheid van de richtlijn te optimaliseren en landelijk draagvlak voor de richtlijn te genereren. Er is door de leden van de NVvH wel gebruik gemaakt van de commentaarronde. De richtlijn werd op 19-12-2023 door de HOVON-CML/MPN-werkgroep inhoudelijk vastgesteld en ter autorisatie gestuurd naar de Nederlandse Vereniging voor Hematologie (NVvH). Het bestuur van de NVvH heeft de richtlijn op geautoriseerd.
In de verschillende fasen van de ontwikkeling van het concept van de richtlijn is zoveel mogelijk rekening gehouden met de implementatie van de richtlijn en de daadwerkelijke uitvoerbaarheid van de aanbevelingen.
Om het gebruik in de dagelijkse praktijk te bevorderen wordt deze richtlijn verspreid onder de professionals van de bij de totstandkoming van deze richtlijn betrokken organisatie(s):
Richtlijncommissie
De richtlijn werd beoordeeld en becommentarieerd door de navolgende leden van de werkgroep:
De leden van de richtlijn commissie hebben verklaard onafhankelijk gehandeld te hebben bij het opstellen van de richtlijn en hebben belangenverklaringen [zie bijlage – Code KNAW/KNMG] ingevuld waarbij is aangegeven welke betrekkingen zij onderhielden met commerciële bedrijven, organisaties of instellingen die in verband staan met het onderwerp van de richtlijn. Een overzicht hiervan kunt u bij het secretariaat van de Nederlandse Vereniging voor Hematologie opvragen.
|
|
Belangen |
|
dr. P.A.W. te Boekhorst, internist-hematoloog/transfusiespecialist |
Novartis, (sprekersgeld) Abbvie, GSK (internationale adviesraad) |
|
dr. S. Kersting, internist-hematoloog |
Geen |
|
dr. R. Raymakers, internist-hematoloog |
Geen |
|
dr. N.P.M. Schaap, internist-hematoloog |
Novartis, Amgen, Janssen (sprekersgeld) |
|
dr. M.A. de Witte, internist-hematoloog |
Novartis, BMS, Sanofi, AOP (sprekersgeld) |
|
dr. M. Wondergem, internist-hematoloog |
Novartis Steering Committee |
SORT grading
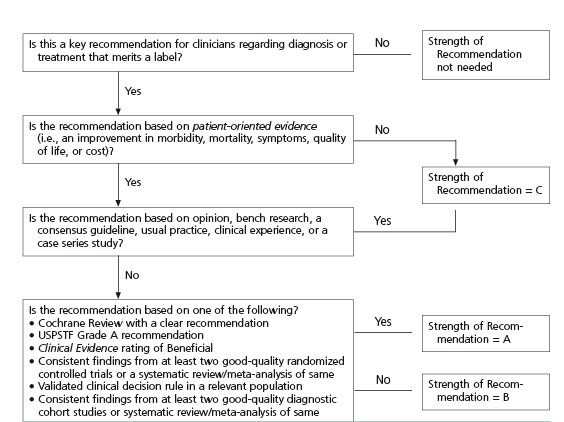
Diagnostiek:
Behandeling:
3. Bij alle patiënten wordt behandeling met trombocyten aggregatieremmers overwogen en gedocumenteerd in het EPD
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn wordt digitaal verspreid onder alle relevante beroepsgroepen. Daarnaast wordt er een toelichting op de richtlijn aangeboden aan het Nederlands tijdschrift voor Hematologie. Ook is de richtlijn te downloaden vanaf de websites van de NVvH (www.hematologienederland.nl) en HOVON (www.hovon.nl)
De patiëntenorganisatie voor deze categorie patiënten is de MPN-stichting. Vertegenwoordigers van de MPN-stichting hebben zitting in de richtlijn werkgroep. Het bestuur is over de conceptrichtlijn geïnformeerd en in staat gesteld te reageren op de inhoud van deze richtlijn.
Welk onderzoek is nodig bij de diagnose PMF?
Aanbevelingen
Welke (prognostische) risico classificaties kunnen worden toegepast bij PMF?
Aanbevelingen
Welk onderverdeling kan gemaakt worden bij MF?
Aanbevelingen
Welke patiënten dienen te worden doorgeleid richting allogene stamceltransplantatie?
Aanbevelingen
Wanneer is gebruik van cytoreductieve therapie geïndiceerd bij pre-PMF en welk behandeldoel moet dan behaald worden?
Aanbevelingen
Wanneer is gebruik van cytoreductieve therapie geïndiceerd bij overte-PMF en welk behandeldoel moet dan behaald worden?
Aanbevelingen
Welke cytoreductieve therapieën zijn geïndiceerd bij PMF?
Aanbevelingen
Welke aanvullende maatregelen zijn geïndiceerd bij PMF?
Aanbevelingen
Welke behandelingen zijn een mogelijkheid bij PMF-geassocieerde splenomegalie?
Aanbevelingen
Welke behandelingen zijn een mogelijkheid bij PMF-geassocieerde anemie?
Aanbevelingen
Wat is het beleid bij een splanchnische veneuze trombose?
Aanbeveling
Aanbevelingen
Tabel 1: MPN-SAF vragenlijst (pdf)
Tabel 2: Diagnostische criteria pre-PMF WHO 2016Khoury 2022
|
Major criteria |
|
|
Minor criteria (laboratorium afwijkingen bij 2 opeenvolgende metingen bepaald): |
|
|
|
|
|
Diagnose pre-PMF |
|
|
*: In de afwezigheid van de drie klonale markers, kan het bepalen van andere somatische mutaties behulpzaam zijn bij het aantonen van klonaliteit (bij voorbeeld: ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc) **: BM-fibrose secundair aan infectie, auto-immuun aandoening, chronisch inflammatoire aandoening, hairy cell leukemie of ander lymfatische neoplasie, gemetastaseerde ziekte, of toxische beenmerg afwijkingen |
Tabel 3: Diagnostische criteria overte PMF WHO 2016Khoury 2022
|
Major criteria |
|
|
Minor criteria (laboratorium afwijkingen bij 2 opeenvolgende metingen bepaald): |
|
|
|
|
|
|
Diagnose overte-PMF |
|
|
*: In de afwezigheid van de drie klonale markers, kan het bepalen van andere somatische mutaties behulpzaam zijn bij het aantonen van klonaliteit (bij voorbeeld: ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc) **: BM-fibrose secundair aan infectie, auto-immuun aandoening, chronisch inflammatoire aandoening, hairy cell leukemie of ander lymfatische neoplasie, gemetastaseerde ziekte, of toxische beenmerg afwijkingen |
Onderbouwing
Om de diagnose te stellen is onderzoek van bloedbeeld en moleculair onderzoek noodzakelijk in combinatie met beenmergonderzoek (Tabel 2 en 3). Er wordt een onderscheid gemaakt tussen pre-PMF en overte-PMF vanwege het verschil in prognose tussen deze twee stadia.Arber 2016 (Zie ook stadiering). Het overgrote deel van de patiënten met PMF heeft constitutionele symptomen zoals vermoeidheid, gewichtsverlies, nachtzweten en koorts. Daarnaast kunnen er ook andere ziekte-gerelateerde klachten zijn zoals concentratiestoornissen, jeuk, botpijn en klachten gerelateerd aan de splenomegalie.Geyer 2014Dit kan bij diagnose en follow-up vastgelegd worden met de MPN-SAF vragenlijst (Tabel 1).Emanuel 2012
Verworven von Willebrand ziekte type II (VvWD) kan voorkomen bij extreme trombocytose maar ook bij lagere trombocyten aantallen.Rottenstreich 2017 Een verminderde ratio (0,7) van von Willbrand ristocetine activiteit ten opzichte van von Willebrand antigeen is daarbij het meest voorspellend.Tiede 2011Aangezien het risico op trombotische complicaties ook bij PMF afhankelijk is van cardiovasculaire risicofactoren dienen deze bij diagnose bepaald te worden.
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de World Health Organisation classificatie, aangevuld met literatuur op basis van expertise van de richtlijnwerkgroep.
Referenties:
Aanbevelingen (SORT B)
Tabel 4: Dynamic International Prognostic Scoring System Plus (DIPSS Plus)Gangat 2011
|
Parameter |
Score |
||
|
Leeftijd >65 jaar Constitutionele symptomen Hb <6,2 mmol/L Leukocytose >25 x 109/L Blasten perifeer bloed ≥1% Trombocytopenie (<100 x109/L) Cytogenetica: complex, +8, -7/7q-, i(17q),5/5q-,12p-, inv(3), of 11q23 rearrangement Erytrocyten transfusie afhankelijkheid |
1 1 1 1 1 1 1 1 |
||
|
Risico indeling DIPSS Plus |
Risicofactoren |
Mediane overall survival (maanden) |
|
|
laag |
0 |
185 |
|
|
intermediair-1 |
1 |
78 |
|
|
intermediair-2 |
2-3 |
35 |
|
|
hoog |
>4 |
16 |
|
Tabel 5: MIPSS70, prognostische score met incorporatie van de moleculaire
diagnostiek Guglielmelli 2018
|
Parameter |
Score |
|
Constitutionele symptomen Hb <6,2 mmol/L Leukocytose >25 x 109/L Blasten perifeer bloed ≥2% Trombocytopenie (<100 x109/L) Beenmerg fibrose graad 2 of graad 3 Hoog-moleculair risico: 1 HMR afwijking* 2 of meer HMR-mutaties* Afwezigheid CALR-type 1 mutatie |
1 1 2 1 2 1 1 2 1 |
|
*ASXL1, SRSF2, IDH1/2, EZH2 |
|
|
Risico indeling MIPSS70 |
Score |
Mediane overall survival (jaren) |
|
laag |
0-1 |
27,7 |
|
intermediair |
2-4 |
7,1 |
|
hoog |
>5 |
2,3 |
Tabel 6: MIPSS70plus v2.0, prognostische score met incorporatie van moleculaire diagnostiek en cytogeneticaTefferi 2018
|
Parameter |
Score |
||
|
– Constitutionele symptomen – Anemie Milde anemie* Ernstige anemie** – Blasten perifeer bloed ≥2% – Mutations Hoog-moleculair risico: – 1 HMR afwijking*** – 2 of meer HMR-mutaties*** Afwezigheid CALR-type 1 mutatie – Karyotype Unfavourable karyotype**** Very high risk karyotype***** |
2
1 2 1
2 3 2
3 4 |
||
|
* Man: Hb >5,6 en <6,8 mmol/L; Vrouw Hb >5,0 en 6,2< mmol/L ** Man: Hb <5,6 mmol/L; Vrouw Hb <5,0 mmol/L ***ASXL1, SRSF2, IDH1/2, EZH2, U2AF1 ****Ieder afwijkend karyotype, behalve: 20q-, 13q-, +9 chromosoom 1 translocatie/duplicatie, -y of geslacht chromosoomafwijking anders dan –y ***** Very high risk (VHR): single of multipele abnormalities of -7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23, of andere autosomale trisomieen maar geen +8/+9 (eg, +21, +19) |
|||
|
Risico indeling MIPSS70-Plus |
Score |
Mediane overall survival (jaren) |
|
|
Zeer laag |
0 |
>10 (niet bereikt in studie) |
|
|
laag |
1-2 |
10,3 |
|
|
intermediair |
3-4 |
7 |
|
|
hoog |
5-8 |
3,5 |
|
|
zeer hoog |
>9 |
1,8 |
|
Onderbouwing
Er zijn diverse score systemen gepubliceerd om de prognose van PMF-patiënten te beoordelen.Barbui 2018 Deze scores zijn over het algemeen gebaseerd op retrospectieve data waarbij soms ook nieuw gediagnosticeerde patiënten geïncludeerd zijn die echter een korte follow-up kennen. Deze score systemen zijn overigens niet gevalideerd voor de patiëntencategorie die ruxolitinib gebruiken of gebruikt hebben. De werkgroep adviseert om de meest recente score systemen te hanteren. De DIPSS-plus score is voor alle PMF-patiënten bruikbaar (Tabel 4), de MIPSS70 en de MIPPS70plus versie 2.0 score voor patiënten van 70 jaar of jonger en kandidaat voor allo- SCT (Tabel 5 respectievelijk Tabel 6). Gangat 2011, Guglielmelli 2018, Alli 2019 Alhoewel de meeste score systemen gevalideerd zijn voor PMF, worden deze in de praktijk ook gebruikt voor post-ET MF en post-PV MF.
Er is een studie waarbij de prognostische score systemen IPSS/DIPSS en DIPSS-plus werden toegepast bij zowel PMF als post-ET en post-PV MF. De overleving was niet significant verschillen tussen deze groepen. Hernández-Boluda 2018
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn richtlijn aangevuld met literatuur op basis van expertise van de richtlijnwerkgroep.
Referenties
Aanbevelingen
Tabel 7: Diagnostische criteria post-PV MFBarosi 2008
|
Vereiste criteria |
|
|
Aanvullende criteria |
|
|
|
|
|
Diagnose post-PV MF |
|
Tabel 8: Diagnostische criteria post-ET MFBarosi 2008
|
Vereiste criteria |
|
|
Aanvullende criteria |
|
|
|
|
|
|
Diagnose post-PV MF |
|
Onderbouwing
Bij pre-PMF is er meestal, net als bij ET sprake van trombocytose. Omdat de prognose van een “echte” ET duidelijk gunstiger is dan van pre-PMF, is het van belang pre-PMF vast te stellen.Swerdlow 2017
Omdat de prognose en symptomatologie van post-PV en post-ET waarschijnlijk gelijk zijn aan die van PMF, is het raadzaam om progressie naar MF vast te stellen. Er zijn aanwijzingen dat het voorkomen van non-driver mutaties bij post-PV en post-ET wel verschilt te opzichte van PMF en daarmee een (negatief) effect kan hebben op de prognose. In de WHO 2016 worden geen aparte diagnostische criteria voor deze diagnoses geformuleerd. Swerdlow 2017 Deze zijn wel in de WHO 2008 gedefinieerd (Tabel 7 en 8). Swerdlow 2008 Deze criteria zijn ook vastgesteld door de International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT).Barosi 2008
Het vastleggen van het risicoprofiel van overte-PMF en de symptomatologie zijn van belang voor het maken van een behandelplan, dat verschilt per risicoprofiel en patiënt kenmerken.Barbui 2018
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de World Health Organisation classificatie, de recente Europese richtlijn aangevuld met literatuur op basis van expertise van de richtlijnwerkgroep.
Referenties
Aanbevelingen
Tabel 9. Myelofibrosis Transplant Scoring system (MTSS)Gagelmann2019
|
Parameter |
Score |
||
|
Leeftijd > 57 jaar Karnofsky score <90% Non-CALR/MPL driver mutatie ASXL1 mutatie Leucocyten aantal >25 x109/L voor allogene stamceltransplantatie Trombocyten aantal <150 x109/L voor allogene stamceltransplantatie HLA-mismatch unrelated donor |
1 1 2 1 1 1 2 |
||
|
Risico indeling |
Score |
5-jaars NRM (%) |
Survival % (5-jaar) |
|
Low |
0-2 |
10 |
83 |
|
Intermediate |
3-4 |
22 |
64 |
|
High |
5 |
36 |
37 |
|
Very High |
6-9 |
57 |
22 |
|
NRM: non-relapse mortality |
|||
Onderbouwing
Allo-SCT blijft vooralsnog de enige curatieve behandeloptie, echter met een transplantatie-gerelateerde mortaliteit van rond de 30-40%. Kerbauy 2007, Kroger 2015 De remissie percentages worden wisselend gerapporteerd, mede veroorzaakt door de verschillende respons criteria die worden gebruikt. Bij de beoordeling of een patiënt met MF in aanmerking komt voor allo-SCT dienen diverse afwegingen gemaakt te worden zoals leeftijd, co-morbiditeit en prognostisch risicoprofiel. De leeftijdsgrens en de intensiteit van conditionering worden lokaal in de transplantatiecentra bepaald. Voor het inschatten van de prognose kunnen diverse score systemen gebruikt worden. Recent zijn 2 risico score systemen gepubliceerd (MIPSS70 en MIPSS70plus v2.0, zie Tabel 5 en 6) die klinische parameters, cytogenetica en moleculaire diagnostiek integreren om tot een prognostische risico inschatting te komen. Het verschil tussen de score systemen is wel of niet gebruik van cytogenetica data. De score systemen zijn ontwikkeld met als doel identificatie van kandidaten voor allo-SCT op basis van analyse van data van 805 PMF-patiënten afkomstig uit twee centra. De data van patiënten werden na analyse gebruikt als ontwikkel -en validatie cohorten. De MIPSS70 onderscheid 3 risicogroepen (laag, intermediair en hoog). De mediane overleving in deze groepen is respectievelijk 27.7, 7.1 en 2.3 jaar. De MIPSS70plus v2.0 score onderscheid 5 groepen (zeer laag, laag, intermediair, hoog en zeer hoog) met de daarbij behoren overall survival van respectievelijk >10 jaar (mediane overleving in studie niet bereikt), 10,3, 7, 3.5 en 1.8 jaar (zie Tabel 5 en 6). Guglielmelli 2018, Tefferi 2018 Alhoewel deze score systemen in een niet dynamisch model ontwikkeld zijn, werd ter validatie wel een dynamisch cohort gebruikt. Deze score systemen kunnen derhalve wel als dynamisch model gebruikt worden.
De werkgroep adviseert bij MF-patiënten die in de MIPSS70 in de hoog risico categorie vallen en MF-patiënten die in de MIPSS70plus in de hoog of zeer hoog risicogroep vallen, allo-SCT te overwegen.
Naast het gebruik van de MIPSS70 en MIPSS70-Plus kan voor een inschatting van het transplantatierisico en outcome gebruik gemaakt worden van de Myelofibrosis Transplantation Scoring System (MTSS score) waarin klinische risicofactoren en moleculaire risicofactoren worden gecombineerd (Tabel 9). Gagelmann 2019
Het stellen van de indicatie kan daarbij losgezien worden van de timing van een allo-SCT. Tijdig overleg met een SCT centrum bij tekenen van acceleratie en/of het ontstaan van additionele, ongunstige non-driver mutaties en/of aanwijzingen voor het ontstaan van ruxolitinib resistentie is geïndiceerd.
Ten aanzien van behandeling met een JAK2 remmer voorafgaand aan, en/of in aansluiting op allo-SCT zijn vooralsnog weinig prospectieve data beschikbaar. Een recente retrospectieve studie liet een voordeel zien van de SCT uitkomsten bij ruxolitinib responsieve patiënten.Kroger 2021, Robin 2021 Reductie van splenomegalie voor allogene SCT door bijvoorbeeld ruxolitinib voorbehandeling, lijkt zeer gewenst.
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van literatuur op basis van expertise van de richtlijnwerkgroep.
Referenties
Aanbevelingen
Onderbouwing
Patiënten met pre-PMF hebben vaak een beeld lijkend op ET. Naar analogie van het verhoogde risico op trombo-embolische complicaties bij ET dat afhankelijk is van zijn voorgeschiedenis, is het advies te starten met cytoreductieve therapie indien er een trombo-embolische complicatie heeft plaatsgevonden. Overwogen kan worden dat ook te doen zodra patiënt ouder dan 60 jaar wordt (bijvoorbeeld op basis van de aanwezigheid van andere risicofactoren). Het doel van de therapie is het verminderen van het risico op (nieuwe) trombo-embolische complicaties door bereiken van normale aantal trombocyten.Barbui 2018 Vaak echter is cytoreductie niet mogelijk door reeds bestaande anemie. VvWD is zeldzaam bij pre-PMF. Om het risico op bloedingen te verkleinen, dient cytoreductieve therapie plaats te vinden bij patiënten met VvWD. Om het bloedingsrisico te verkleinen is normalisatie van trombocyten aantal niet per se noodzakelijk, zolang er complete remissie is van VvWD. Ook pre-PMF gerelateerde syptomen of symptomatische splenomegalie kunnen een indicatie zijn voor cytoreductieve therapie, mits de cytopenie dit toelaat.
Leucocytose is geassocieerd met een verhoogd risico op het ontstaan van trombo-embolische complicaties bij PV. Naar analogie hiervan kan overwogen worden om bij leucocytose te starten met cytoreductie.
Deze aanbevelingen zijn gelijk aan de aanbevelingen die bij overte-PMF gehanteerd worden.
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn en expertise van de richtlijnwerkgroep.
Referentie
Aanbevelingen
Onderbouwing
Voor patiënten met DIPSS-plus laag of intermediair-1 risico PMF is geen behandeling die het ziektebeloop kan beïnvloeden. Een recent onderzoek laat zien dat het verminderen van de JAK2 allelic burden (in deze studie d.m.v. ruxolitinib), mogelijk een gunstig effect heeft op het ziekte beloop.Harrison 2023 Het primaire eindpunt van deze studie was echter niet bedoeld om dat aan te tonen. Behandeling is daarom vooralsnog alleen geïndiceerd indien er klachten zijn (constitutionele symptomen) of indien door de myeloproliferatie klachten of complicaties te verwachten zijn (zie voor onderbouwing ook uitgangsvraag 5, cytoreductieve bij pre-PMF). Barbui 2018 Er zijn onvoldoende klinische data beschikbaar om een behandeling met cytoreductieve therapie bij PMF patiënten met een trombocyten aantal <400 x109/L en een door gemaakte trombo-embolische complicatie te rechtvaardigen.
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn en expertise van de richtlijn werkgroep.
Referentie
Aanbevelingen
Tabel 10: aandachtspunten bij start ruxolitinib
|
Medicatie niet abrupt staken maar de dosis in 2 weken verminderen ter preventie van “withdrawal syndroom” |
|
Geneesmiddelen interactie met sterke/matige CYP3A4 remmers of tweevoudige remmers van CYP2C9 en CYP3A4. Voorbeelden: claritromycine, ciprofloxacine, itraconazol, ketoconazol, cimetidine, verapamil |
|
Gezien het effect van ruxolitinib op de cellulaire afweer moet voorafgaand aan het starten van ruxolitinib overwogen worden of screening op tuberculose zinvol is |
|
Ondanks toename van het infectierisico bij ruxolitinib gebruik is standaard antimicrobiële of virale profylaxe niet geïndiceerd. Patiënt dient te worden voorgelicht ten aanzien van verhoogd risico op herpes zoster reactivatie. Overweeg recombinant herpes zoster vaccin (Shingrix®) indien vergoeding geregeld |
Tabel 11: definities ruxolitinib refractairiteit, recidief/progressie en intolerantie
|
Ruxolitinib |
|
|
Refractairiteit |
Ruxolitinib therapie > 3 mnd met <10% miltvolume reductie of <30% milt grootte tov baseline |
|
Recidief/progressie ziekte |
Ruxolitinib therapie > 3 mnd met verlies van milt respons |
|
Intolerantie |
Ruxolitinib therapie > 28 dagen met ontstaan van erythocyten transfusie-afhankelijkheid (> 2 E/maand gedurende 2 mnd), of > gr 3 trombopenie en/of anemie en/of bloeding en/of hematoom |
Tabel 12 aandachtspunten bij start fedratinib
|
De behandelindicatie voor fedratinib behoort te worden vastgelegd in een multidisciplinair overleg |
|
Vóór de start van de behandeling en tijdens de behandeling moet de thiaminespiegel worden gecorrigeerd indien deze te laag is. Tijdens de behandeling moet de thiaminespiegel periodiek worden bepaald (bijv. maandelijks gedurende de eerste 3 maanden en daarna om de 3 maanden) en indien klinisch geïndiceerd |
|
Als gelijktijdig gebruik van sterke CYP3A4-remmers niet kan worden vermeden, moet de dosis van Fedratinib worden verlaagd tot 200 mg |
|
Ten aanzien van gastro-intestinale bijwerkingen kunnen de volgende maatregelen worden genomen:
|
|
De effectiviteit van de fedratinib behandeling dient na 3 maanden beoordeeld te worden om te kunnen beslissen of de behandeling voortgezet kan worden. Respons wordt dan gedefinieerd als een afname van miltafmeting >10 % met afname van klachten veroorzaakt door de splenomegalie en/of afname van ziekte gerelateerde klachten. Dit laatste behoort te worden vastgelegd door middel van de MPN score lijst waarbij een verbetering van 10% of meer moet worden bereikt t.o.v. uitgangsituatie |
Onderbouwing
Hydroxycarbamide is van oudsher het meest gebruikte middel bij MPN en een goede keus bij laag risico PMF met behandelindicatie. In geval van een vroege fase van (pre-) PMF kan echter ook een behandeling met gepegyleerd interferon α 2a overwogen. Er is geen wetenschappelijke onderbouwing welk middel de voorkeur heeft mogelijk. Bij PV en ook ET zijn gunstige effecten van gepegyleerd interferon α 2a op de ‘allelic’ burden beschreven. Mascarenhas 2023 Mogelijk treedt dit effect ook op bij de behandeling van PMF. Momenteel is nog onduidelijk in hoeverre het ziektebeloop hierdoor beïnvloed wordt. Op basis van deze gegevens is vooralsnog geen duidelijke voorkeur aan te geven welke behandeling de voorkeur heeft.
Bij de keuze tussen de behandelopties dient een mogelijke hogere incidentie van non-melanoma huidmaligniteiten na langdurig hydroxycarbamide gebruik in de overweging te worden meegenomen. Gezien het bijwerkingen profiel van gepegyleerd interferon α 2a kan een startdosering van 45 microgram/week s.c. verkozen worden boven een hogere startdosering. Inmiddels heeft de European Medicines Agency (EMA) ropeginterferon alfa-2b (Besremi®) goedgekeurd als monotherapie voor gebruik bij volwassenen voor de behandeling van PV zonder symptomatische splenomegalie. Dit middel wordt echter niet voor de behandeling van PMF geregistreerd. Ruxolitinib kan worden voorgeschreven aan patiënten met symptomatische splenomegalie en/of ziekte gerelateerde symptomen. Indien alleen symptomatische splenomegalie op de voorgrond staat, kan in eerste instantie het effect van behandeling met hydroxycarbamide afgewacht worden, bij falen kan overgegaan worden tot ruxolitinib behandeling. Om in dat geval een maximaal effect te bereiken wordt geadviseerd te starten met een hogere dosering ruxolitinib en daarna, afhankelijk van trombocytenwaarde, de dosering eventueel aan te passen. Anemie vormt op zich geen contra-indicatie voor het starten van therapie of het aanpassen van de dosering. Ingeval van ernstige ziekte-gerelateerde symptomen is de eerste keus ruxolitinib.Barbui 2018 Bij twijfel over het starten van ruxolitinib op deze indicatie kan een proefbehandeling van 6-8 weken overwogen worden. De keuze voor een van de mogelijke cytoreductieve behandelingen dient afhankelijk van de individuele patiënt en ziekte kenmerken gemaakt te worden. Het gebruik van fedratinib in de 1e lijn lijkt op basis van de gepubliceerde studies geen duidelijke meerwaarde te hebben (expert opinion).
In geval van ineffectiviteit, intolerantie of verlies van effectiviteit van ruxolitinib kan overwogen worden fedratinib (JAK2/FLT3 remmer) in te zetten (Tabel 11). De te verwachten effectiviteit in de vorm van miltrespons ligt rond de 30%. Effect op ziekte-gerelateerde symptomen ligt eveneens rond de 30%. Harrison 2019 Indicatiestelling dient vastgelegd te worden in een multidisciplinair overleg (MDO). Respons beoordeling na 3 maanden behandeling is een aanvullende voorwaarde voor de vergoeding van een voortgezette behandeling (Tabel 12). Gezien het mogelijk optreden van gastro-intestinale toxiciteit dient hier speciale aandacht aan geschonken te worden. In de FREEDOM trial werd gedurende de 1e cyclus van fedratinib behandeling werd WHO graad 1 en 2 toxiciteit gezien in de vorm van diarree (21%), nausea (21%) en braken (12%). Er werd geen graad 3 of 4 toxiciteit gerapporteerd van deze bijwerkingen. Dit percentage verminderde sterk tijdens de navolgende behandelcycli. Middels anti-emetica en anti-diarree middelen was de toxiciteit goed beheersbaar. Thiamine spiegels kunnen bij daling (weinig voorkomend) door orale suppletie worden opgevangen.Gupta 2020
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn en expertise van de richtlijnwerkgroep.
Referenties
Aanbevelingen
Onderbouwing
Vanwege het verhoogde risico op trombo-embolische complicaties dienen alle patiënten trombocyten aggregatieremming (TAR) te krijgen, tenzij hiervoor een contra-indicatie is. Indien er sprake is van klinische bloedingsneiging en/of VvWD (ratio van von Willbrand ristocetine activiteit ten opzichte van von Willebrand antigeen <0,7) dient dit eerst met cytoreductieve therapie behandeld te worden. In geval van VvWD en een ristocetine activiteit <0,35% is adviseert de werkgroep om trombocyten aggregatie remming te staken en te hervatten na effectieve cytoreductieve therapie. Beperkte data verkregen via een studie bij patiënten met essentiële trombocytose (ET) laten zien dat tweemaal daags doseren van TAR effectiever kan zijn in vergelijking met eenmaal daags doseren. Rocca 2020 Meerdaags doseren echter kan wel leiden tot meer bijwerkingen. De werkgroep adviseert om deze reden om vooralsnog niet om standaard tot een tweemaal daagse doseringsregime over te gaan. Daarnaast zijn er beperkte data die laten zien dat een avond dosering van TAR effectiever kan zijn dan een ochtend dosering. Deze data zijn gebaseerd op in vitro onderzoek verricht bij gezonde vrijwilligers (n=12) zonder klinische eindpunten. Racca 2019 Op grond van deze data is de werkgroep van mening dat overwogen kan worden een avond dosering te adviseren in plaats van een ochtend dosering. Bij patiënten met een indicatie voor clopidogrel moet een inschatting gemaakt worden of dubbeltherapie met acetylsalicylzuur of carbasalaatcalcium opweegt tegen het bloedingsrisico van deze combinatie. Er zijn onvoldoende gegevens beschikbaar over clopidogrel monotherapie. Patiënten met PMF hebben een verhoogd risico op het krijgen van jicht of uraatsteen. Bij start van cytoreductieve therapie dient afgewogen te worden of profylaxe geïndiceerd is. Aangezien patiënten vaak algemene klachten hebben en behoudens allo-SCT er geen behandelingen zijn die het ziektebeloop beïnvloedden is goede supportive care van belang.
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de expertise van de richtlijnwerkgroep.
Referenties
Aanbevelingen (SORT C)
Onderbouwing
Bij laag of intermediair risico PMF met splenomegalie is hydroxycarbamide een goede behandeloptie, tenzij er symptomatologie is die ruxolitinib behandeling rechtvaardigt. Bij intermediair-2 of hoog risico PMF is ruxolitinib eerste keus bij splenomegalie. In geval van ruxolitinib refractairiteit/ineffectiviteit of intolerantie kan fedratinib overwogen worden. Harrison 2019 Splenectomie is een optie als patiënten resistent of intolerant zijn tegen de medicamenteuze behandelopties. Barbui 2018 Splenectomie zo mogelijk verrichten voordat trombocyten <50 x109/l zijn vanwege bloedingsrisico bij operatie.
Miltbestraling is ook een optie bij mechanische bezwaren van splenomegalie. Gezien de hoge kans op pancytopenie moet worden gekozen voor een lage fractiedosis. De mediane responsduur is ongeveer 6 maanden. Gezien de lage frequentie van zowel splenectomie als milt bestraling bij PMF, het ontbreken van een eenduidige indicatie en hoge kans op complicaties of pancytopenie is overleg met consulterend behandelcentrum aanbevolen.
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn en expertise van de richtlijnwerkgroep.
Referentie
Aanbevelingen (SORT C)
Onderbouwing
Behandeling van PMF-geassocieerde anemie is mogelijk met erythropoëtine, danazol, corticosteroiden en immuunmodulerende geneesmiddelen (IMiDs). Bij de keuze voor een specifieke behandeling dient de toxiciteit van de behandeling en individuele patiënt kenmerken te worden meegewogen.Barbui2018 De kans op een respons met erythropoëtine is bij een laag serum erytropoëtine ongeveer 50% bij een normaal serum erytropoëtine veel lager.Hernandez-Boluda2017 Risico van behandeling is verergering van splenomegalie. Behandeling met danazol geeft bij ongeveer 30% respons na 3-6 maanden, waarvan bij de helft een duurzame respons met 100-200 mg/dag onderhoudsbehandeling.Cervantes2015 Bij het voorschrijven van dit middel aan vrouwen dient het risico op virilisatie overwogen te worden, alhoewel dit bij het gebruik van lagere doseringen geen probleem lijkt te zijn. Er zijn positieve resultaten bij lenalidomide, al dan niet in combinatie met corticosteroïden. Dit middel kan ‘off label’ worden voorgeschreven voor deze indicatie.Chihara 2016
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn en expertise van de richtlijnwerkgroep.
Referentie
Aanbeveling (SORT C)
Onderbouwing
Splanchnische veneuze trombose (SVT) is een veneuze trombo-embolie van een of meer van de abdominale venen(porta, lienalis, mesenterica, of supra-hepatische of intra-hepatische (Budd-Chiari syndroom) venen) en kan ernstige bloedingen als gevolg van portale hypertensie, splenomegalie en varices veroorzaken. Indien er sprake is van PV volgens de WHO criteria is er een indicatie voor cytoreductieve therapie op basis van het optreden van een trombo-embolische complicatie.Barbui 2018 In principe is er bij alle patiënten met SVT een indicatie voor therapeutische antistolling, maar dit dient afgewogen te worden tegen het risico op bloedingen.Riva 2012, Candeloro 2022 Bij SVT in het kader van een MPN heeft levenslange therapeutische antistolling (tenzij contra-indcatie) de voorkeur boven teruggaan op Acetylsalicylzuur 80 mg/Carbasalaatcalcium 100 mg/ dag na de initiële periode van antistolling; een systematische review laat zien dat antistolling, in combinatie met cytoreductieve therapie, het recidief risico na trombose het meest effectief verlaagd.Hamulyak 2021 Er zijn onvoldoende gegevens beschikbaar om algemene aanbevelingen te doen over het toevoegen van trombocyten aggregatieremming naast therapeutische antistolling, aangezien vooral het bloedingsrisico hierdoor wordt vergroot. Bij de individuele patiënt kan dit overwogen worden in geval van een recidief trombose onder adequate antistollingstherapie en met afweging van risicofactoren voor bloeding. Ten aanzien van het gebruik van DOACs i.p.v. vitamine K antagonisten of LMWH kan het volgende gesteld worden: op basis van niet-gerandomiseerde studies lijken DOAC even effectief en veilig in het verlagen van het recidief risico na trombose als andere types antistolling (LMWH, VKA) en kunnen dus worden overwogen.Ageno 2022 Een duidelijke voorkeur voor het type antistolling is niet te geven, en dient per patiënt te worden afgewogen mede in afweging van hepatologische factoren. Ageno 2022,Candeloro 2022
Zoekverantwoording
Er is geen systematische literatuuranalyse verricht, maar gebruik gemaakt van de recente Europese richtlijn, een expert opinion artikel over dit onderwerp en expertise van de richtlijnwerkgroep.
Referenties
© 2022. Alle rechten voorbehouden
Nederlandse Vereniging voor Hematologie (NVvH)
Uiterlijk in 2027 bepaalt dr. P.A.W te Boekhorst en het bestuur van de NVvH of deze richtlijn of module nog actueel is. Zo nodig wordt een nieuwe werkgroep geïnstalleerd om de richtlijn te herzien. De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten. Dr. P.A.W. te Boekhorst is als houder van deze richtlijn de eerstverantwoordelijke wat betreft de actualiteit van deze richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de eerst-verantwoordelijke over relevante ontwikkelingen binnen hun vakgebied.