Terug naar het richtlijnenoverzicht
Dit document beoogt het beschikbaar maken van richtlijnen voor de behandeling en begeleiding van patiënten met sikkelcelziekte. Het document is geschreven door verschillende disciplines verenigd in de subwerkgroep hemoglobinopathie behandelaren. In deze landelijke werkgroep hemoglobinopathie behandelaren (LWHB) zijn internist-hematologen, kinderarts-hematologen, klinisch genetici, laboratoriumspecialisten en gespecialiseerde verpleegkundigen vertegenwoordigd. Ook is er regelmatig contact met de patiëntenverenigingen Oscar en IXL.
Uiterlijk vijf jaar na verschijnen van de definitieve richtlijn zal worden beoordeeld of herziening nodig is. Wanneer ontwikkelingen in de toekomst het eerder noodzakelijk maken zal een herzieningsprocedure worden gestart.
Doel is een multidisciplinaire behandelrichtlijn voor kinderen en volwassenen met sikkelcelziekte te ontwikkelen.
De richtlijn beoogt een praktisch handvat te bieden aan internisten, internist-hematologen, kinderartsen en kinderarts-hematologen.
Initiatief nemende organisatie:
Werkgroep benigne hematologie van de Nederlandse Vereniging voor Hematologie – subwerkgroep Hemoglobinopathie Behandelaren.
Overige betrokken organisaties: Nederlandse Vereniging voor Kindergeneeskunde, sectie Kinderhematologie en patiëntenorganisatie OSCAR.
Te autoriseren door: Nederlandse Vereniging voor Hematologie
Werkgroepleden richtlijn:
Alle werkgroepleden hebben verklaard onafhankelijk gehandeld te hebben bij het opstellen van de richtlijn. Aan de werkgroepleden is gevraagd een belangenverklaring in te vullen, waarin ze hun banden met de farmaceutische industrie aangeven
De richtlijn is tot stand gekomen met medewerking van alle werkgroepleden benoemd in de lijst van de richtlijn die allen zitting hebben in de landelijke werkgroep hemoglobinopathie behandelaren (LWHB). De richtlijn is tot stand gekomen middels het raadplegen van de recente literatuur en het raadplegen van andere richtlijnen.
Methode ontwikkeling
Voor het vinden van relevante informatie is gebruik gemaakt van Pubmed en andere internationale richtlijnen.
Werkwijze
Er werd voor alle afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in de elektronische databases van Pubmed. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen en bestaande internationale richtlijnen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroep leden selecteerden de via de zoekactie gevonden artikelen op basis van op voorhand opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekactie of gebruikte trefwoorden van de zoekactie en de gehanteerde selectiecriteria zijn te vinden in de module van de desbetreffende uitgangsvraag.
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn is/wordt digitaal verspreid onder alle relevante beroepsgroepen. Daarnaast is/wordt er een toelichting op de richtlijn aangeboden aan het tijdschrift voor Hematologie. Ook is de richtlijn te downloaden vanaf de website van de Nederlandse Vereniging voor Hematologie (NVvH): www.hematologienederland.nl . In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn wordt digitaal verspreid onder alle relevante beroepsgroepen. Daarnaast is/wordt er een toelichting op de richtlijn aangeboden aan het Nederlands tijdschrift voor Hematologie. Ook is de richtlijn te downloaden vanaf de website www.hematologienederland.nl
In deze richtlijn worden de patiënten vertegenwoordigd door een afgevaardigde van OSCAR Nederland.
Uitgangsvraag
Welke onderzoeken zijn in welke frequentie nodig om orgaanschade bij patiënten te detecteren?
Aanbeveling
Het schema is een aanbeveling in welke frequentie screeningsonderzoeken moeten worden gedaan. Daarbij is een onderscheid gemaakt tussen het eerste consult en poliklinisch vervolgconsult. De frequentie van de onderzoeken kan aangepast worden indien sprake is van orgaanschade.
|
Basisdiagnostiek eerste consult (geldt niet voor baby’s met afwijkende hielprik) |
|
Hb, MCV, leukocyten, trombocyten, reticulocyten Hb typering (HPLC), alfa-thalassemie onderzoek, DNA-diagnostiek Uitgebreide bloedgroep typering (AB0, Rh volledig uitgetypeerd, K, Fy(a), Jk(a), Jk(b) en MNS-systeem (M, S en s antigenen) |
|
Bilirubine (direct/totaal), LDH, kreatinine, ASAT, ALAT , AF en γGT, ferritine/transferrinesaturatie (TSAT), NT-proBNP |
|
Parvovirus B19 virusserologie Hepatitis B, C HIV |
|
Genetische counseling (bij voorkeur verwijzing naar klinisch geneticus) |
|
Basisdiagnostiek eerste consulten voor baby’s met een afwijkende hielprik |
|
|
Hb, MCV, leukocyten, trombocyten, reticulocyten |
Diagnose, 6 en 12 maanden |
|
Hb typering (HPLC) |
Diagnose en 12 maanden |
|
Uitgebreide bloedgroep typering |
12 maanden |
|
alfa-thalassemie onderzoek, DNA-diagnostiek |
Diagnose of 6 maanden |
|
Genetische counseling (bij voorkeur verwijzing naar klinisch geneticus) |
In het 1e jaar |
|
Vervolgonderzoek |
Eerste keer op leeftijd van |
Frequentie |
|
Hb, MCV, leukocyten, evt. differentiatie, trombocyten, reticulocyten |
1 jaar |
1 a 2x/jaar |
|
Bilirubine (direct/total), LDH, ferritine,transferrine verzadiging (TSAT) |
1 jaar |
1x/jaar |
|
foliumzuur, vitamine B12, vitamine D |
1 jaar |
1x/jaar |
|
ALAT, ASAT, AF, γGT, glucose, NT-proBNP |
1 jaar |
1x/jaar |
|
Kreatinine |
1 jaar |
2x/jaar |
|
Parvo B19 serologie |
5 jaar |
1x/jaar (indien negatief en polytransfusé) |
|
G6PD-deficientie |
1 jaar/bij diagnose |
eenmalig |
|
Bloeddruk |
5 jaar |
1x/jaar |
|
O2 saturatie |
5 jaar |
Op indicatie (o.i.) |
|
Transcraniële doppler (TCD) bij HbSS/ HbSβ0 Overweeg bij HbSD, HbSE ea genotypes die eveneens een hemolytisch fenotype hebben. |
2 jaar
2 jaar |
2-16 jaar 1 a 2x/jaar
2-16 jaar 1 a 2x/jaar |
|
MRI/MRA hersenen |
± 8 jaar (zonder sedatie) |
Op indicatie (slechte school resultaten, beperkte cognitie) Volwassenen op zijn minst 1x, nadien op indicatie Indien stille infarcten op de MRI aangetoond, dan jaarlijks of 1x/2 jaar herhalen |
|
ECG |
Op indicatie |
Op indicatie |
|
Echo cor (1) |
15 jaar |
1x/3 jaar, bij afwijkingen frequenter |
|
Echo abdomen |
Eenmalig op 15 jaar |
Op indicatie |
|
Longfunctie |
Op indicatie |
Op indicatie |
|
Oogheelkundig onderzoek (2) |
12 jaar |
1x/3 jaar bij afwijkingen of in geval van HbSC frequenter |
|
Urineportie op microalbumine , microalbumine/kreat ratio |
Vanaf 10 jaar |
1x/jaar |
1 Echocardiografie (dimensies ventriculi en atria, diastolische functie, sPAP druk, LV en RV ejectiefractie, pieksnelheid tricuspidalisklep (TRV) in verband met pulmonale hypertensie).
2 Consult oogheelkunde voor fundoscopie.
|
Polytransfusee |
|
|
Bloedbeeld |
4x/jaar |
|
HbS% |
Minimaal 4x per jaar |
|
Ferritine, TSAT |
4x/jaar |
|
IgG parvovirus B19 |
1x/jaar indien negatief |
|
Genotypering op bloedgroepgenen wordt aangeraden |
Eenmalig bij Sanquin Amsterdam |
|
Bij chelatietherapie |
|
|
Bloedbeeld |
4x/jaar |
|
Ferritine |
4x/jaar bij ijzerstapeling na transfusies 2x/jaar indien geen transfusies of ijzerstapeling |
|
Zink, Natrium, Kalium, geïoniseerd Calcium, Fosfaat, vitamine D en glucose |
1x/jaar |
|
Kreatinine |
4x/jaar |
|
Urine; eiwit, microalbumine/kreat ratio (bij positieve test, herhalen in ochtendportie), glucose |
1x/jaar |
|
ASAT, ALAT, AF, γGT, bilirubine |
4x/jaar |
|
FT4, TSH, LH, FSH, IGF1, testosteron |
1x/ 2 jaar indien inadequate ontijzering |
|
Audiogram/ICC oogheelkunde |
1x/jaar bij chelatietherapie |
|
MRI T2* hart en lever |
Bij ijzerstapeling of >20 transfusies in het verleden, indien afwijkend, 1x/2 jaar |
|
Botdensitometrie (DEXA scan) |
Bij alle patiënten vanaf 18 jaar eenmalig en daarna 1x/5 jaar |
Achtergrondinformatie (onderbouwing)
De levensverwachting van patiënten met sikkelcelziekte is beperkt als gevolg van orgaanschade. Door systematische screening kan orgaanschade in een vroeg stadium worden gediagnosticeerd en kunnen preventieve maatregelen worden genomen. De incidentie van orgaanschade is in de meeste gevallen niet gerelateerd aan de klinische presentatie. Dit vormt de reden dat bij alle patiënten met sikkelcelziekte systematische screening op secundaire orgaanschade moet plaatsvinden.
Ter preventie van neurologische complicaties wordt geadviseerd om bij kinderen met HbSS/ HbSβ0 thalassemie en andere genotypen met hemolytisch fenotype minimaal jaarlijks een TCD te verrichten vanaf de leeftijd van 2 jaar tot en met 16e jaar.
Gezien de hoge prevalentie van stille cerebrale infarcten bij kinderen met HbSS of HbSβ0 thalassemie en hun associatie met cognitieve stoornissen, slechte schoolprestaties en toekomstige cerebrale infarcten wordt door het ASH-panel aanbevolen om tenminste eenmaal een MRI/MRA-hersenen, zonder sedatie, te verrichten om stille herseninfarcten bij kinderen in de vroege schoolgaande leeftijd (± 8 jaar) te detecteren. En dit minimaal 1x op de volwassen leeftijd te herhalen.
De belangrijkste cardiovasculaire aandoeningen bij sikkelcelziekte zijn diastolische dysfunctie, pulmonale hypertensie en linker ventrikel dysfunctie. Amerikaanse aanbevelingen voor screening is een echocardiografie wanneer er klachten zijn die wijzen op pulmonale hypertensie. Europese experts bevelen aan om 1x per 3 jaar een echocardiografie te verrichten.
Screening voor retinopathie, bij alle genotypen, wordt 2-jaarlijks geadviseerd vanaf de leeftijd van 10 jaar. Het bewijs voor deze aanbeveling is van lage kwaliteit. Voorstel, o.b.v. literatuur, om te starten op leeftijd 12 jaar en 3-jaarlijks follow up. Bij afwijkende bevindingen en/of visusklachten uiteraard eerder. Bij het genotype HbSC wordt meer frequente screening aanbevolen, zie hoofdstuk HbSC.
Zoekverantwoording
Er is gebruikt gemaakt van bestaande richtlijnen en literatuur.
Uitgangsvraag
Wat is voor patiënten met een vaso-occlusieve crise (VOC) de optimale behandeling?
Aanbeveling
De hoeksteen van de behandeling is adequate pijnstilling met paracetamol, non-steroïd anti inflammatory drugs (NSAID’s) en/of opiaten. Hieraan kunnen zo nodig andere pijnstillers worden toegevoegd, zoals clonidine en ketamine. Deze kunnen per os, subcutaan, rectaal of intraveneus worden toegediend. Geadviseerd wordt om pijnstilling te starten volgens een geïndividualiseerd behandelplan (evt. in overleg met pijnbehandelteam). Indien de patiënt nog geen behandelplan heeft, wordt geadviseerd te starten met een bolus morfine van 0.1 mg/kg iedere 20-30 minuten tot pijnscore < 7, waarna verdere pijnstilling liefst middels patiënt controlled analgesia (PCA). PCA is in het algemeen ongeschikt voor kinderen < 6 jaar of patiënten met een verstandelijke beperking. De ervaring leert dat kinderen met sikkelcelziekte onder de leeftijd van 10 a 12 jaar moeite hebben om dit adequaat toe te passen. Bij hen wordt gebruik gemaakt van een continue morfine pomp (dosering: 10-30 ug/kg/u). Indien nog geen mogelijkheid tot iv behandeling met opiaten, dan kan ter overbrugging een fentanyl neusspray (dosering naar gewicht) worden overwogen. Het gebruik van pethidine wordt afgeraden in verband met neurotoxiciteit. Bij onvoldoende pijnstilling onder maximale doseringen opiaten, kan behandeling met clonidine en zo nodig ketamine overwogen worden (in samenspraak met het lokale pijnbehandelteam). Een ongecompliceerde pijncrise vormt geen indicatie voor erytrocyten transfusie of erytroferese/wisseltransfusie. Voor indicaties voor bloedtransfusie verwijzen we naar de paragraaf bloedtransfusie.
Er bestaan geen prospectieve studies naar het effect van hyperhydratie op de duur van een VOC. In veel centra wordt 3 liter per dag per os of intraveneus met NaCl 0.65% geadviseerd bij volwassen patiënten gedurende de eerste 72 uur, tenzij dit beleid eerder tot overvulling heeft geleid. Er zijn echter ook centra waar geen hyperhydratie wordt toegepast. Dan worden patiënten gestimuleerd en gemonitord om goed te drinken. Voor pediatrische patiënten werd voorheen 2-3 l/m2 geadviseerd met een maximum van 3l/dag gedurende maximaal 72 uur. Bij kinderen (en zeker bij diegene waarbij eerder overvulling werd gezien) kan worden volstaan met het waarborgen van het totaal onderhoudsvocht iv op basis van het gewicht.
Zuurstof dient alleen toegediend te worden als de saturatie <95% is. Er is onvoldoende bewijs voor hoog-risico profylaxe (1dd 5700 IU laagmoleculair gewichtsheparine). Bij opgenomen patiënten geldt het standaard trombose profylaxe beleid.
| Aanbeveling |
Score |
|
Bij een sikkelcelpatiënt met een VOC dient binnen 30 min na binnenkomst adequate pijnstilling gestart te worden |
A1 |
|
Bij een VOC wordt geadviseerd direct met paracetamol, NSAID’s en morfine te starten |
B2 |
|
Er is geen bewijs voor de waarde van hyperhydratie bij volwassenen met een VOC maar orale of intraveneuze hyperhydratie met NaCl 0.65% gedurende 72 uur kan overwogen worden, tenzij er eerder sprake was van overvulling of er sprake is van een acuut chest syndroom. Bij kinderen met een VOC (en zeker bij diegenen die eerder overvulling hebben getoond) kan worden volstaan met het waarborgen van het totaal onderhoudsvocht iv op basis van het gewicht. |
C3
B2 |
Achtergrondinformatie (onderbouwing)
Ruim 15% van de patiënten met sikkelcelziekte heeft meer dan 1 vaso-occlusieve crise per jaar waarvoor opname noodzakelijk is. Daarentegen heeft een kwart van de patiënten nooit een crise waarvoor opname noodzakelijk is. Het is van belang te onderkennen dat een VOC een zeer pijnlijke acute complicatie is waarvoor in de meeste gevallen behandeling met opiaten noodzakelijk is. Verschillende gerandomiseerde en observationele studies rechtvaardigen het gebruik van NSAID’s en opiaten in de acute fase van een VOC. Gezien het verminderd concentrerend vermogen van de nieren bij patiënten met sikkelcelziekte en de noodzaak van een goede hydratietoestand wordt hyperhydratie met NaCL 0,65% geadviseerd gedurende 72 uur. Bij kinderen (en zeker bij diegene waarbij eerder sprake was van overvulling) kan worden volstaan met het waarborgen van het totaal onderhoudsvocht iv op basis van het gewicht.
Aangezien in een studie uit 2021 is gebleken dat hyperhydratie vaak (bij 21% van de patiënten) aanleiding geeft tot overvulling als gevolg van een verminderde hartfunctie, waarvoor behandeling nodig is en waardoor 2 dagen verlenging van de opnameduur optreedt, is het advies om in elk geval volwassenen die eerder overvulling hebben getoond en kinderen met SCZ een normale hoeveelheid iv vocht voor te schrijven.
Gerandomiseerde studies naar het nut en de bijwerkingen van hyperhydratie ontbreken echter. Bij patiënten die zich presenteren met een acuut chest syndroom wordt een restrictief vochtbeleid geadviseerd (1,5 liter/24 uur).
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen.
Uitgangsvraag
Welke vorm van anticonceptie wordt geadviseerd bij een patiënt met sikkelcelziekte?
Aanbeveling
Oestrogeen houdende orale anticonceptie is een relatieve contra-indicatie in verband met de verhoogde tromboseneiging bij patiënten met sikkelcelziekte.
Voor anticonceptie wordt progestagene anticonceptie (depot preparaat) of een levonorgestrel-houdend intra uterine device (IUD) geadviseerd.
|
Aanbeveling |
Score |
|
Progestagene anticonceptie of een levonorgestrel-houdend IUD |
A3 |
Achtergrondinformatie (onderbouwing)
Patiënten met sikkelcelziekte hebben een cumulatieve kans van 11,3% op de ontwikkeling van veneuze trombose. Het risico op trombose neemt toe met de jaren: de cumulatieve trombose incidentie kan oplopen tot 17,1% op het 40stelevensjaar bij patiënten met een ernstige vorm van sikkelcelziekte (HbSS en >3 opnames per jaar). Deze incidentie is aanzienlijk hoger in vergelijking met patiënten zonder sikkelcelziekte. Gezien dit verhoogde risico bestaat er een relatieve contra-indicatie voor de oestrogeen bevattende orale anticonceptie. In een cohortonderzoek werd tevens aangetoond dat oestrogeen bevattende orale anticonceptie leidt tot een 4x hoger risico op een ischemisch CVA leidt.
De levonorgestrel hormoon spiraal (Mirena) geeft geen verhoogd risico op VTE. De pil met alleen desogestrel 75 ug (minipil) lijkt nauwelijks een verhoogd risico op VTE te geven. De gecombineerde anticonceptiepil verhoogd het risico op VTE waarbij pillen met levonorgestrel relatief het veiligst zijn. Een recente retrospectieve analyse onder 7173 orale anticonceptie gebruikers met sikkelcelziekte, werd er geen verschil gevonden tussen gebruikers van combinatie orale anticonceptie en progestagene anticocnpetie ten aanzien van eht voorkomen van trombo-embolische events.
Pillen met andere oestrogenen, te weten gestodeen, desogestrel, cyproteronacetaat en drospirenon, verhogen het risico aanmerkelijk meer. Het gebruik van een implantatiestaafjes (Implanon) lijkt het risico niet te verhogen maar er zijn onvoldoende gegevens om een relevante risicostijging uit te sluiten. De prikpil, de hormoonpleister en een vaginale ring geven ook een verhoogd tromboserisico.
Progestagenen anticonceptie kunnen voorgeschreven worden aan patiënten met sikkelcelziekte. Echter, het voordeel van lage dosis combinatie middelen en intra uteriene spiralen wegen in het algemeen wel op tegen de risico’s van ongewenste zwangerschap in patiënten met sikkelcelziekte.
Het toegenomen tromboserisico van het gebruik van combinatie preparaten in de algemene populatie (verdubbeling van het aantal veneuze tromboses) moet besproken worden met de patiënt. Er zijn geen studies die aantonen dat het gebruik van progestagenen anticonceptie tot minder trombose leidt in patiënten met sikkelcelziekte, wel in de algehele bevolking.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen.
Uitgangsvraag
Aanbeveling
Acute erytroferese of wisseltransfusie is geïndiceerd bij ernstige sikkelcelziekte gerelateerde complicaties zoals: acute chest syndroom met hypoxie, sepsis/multi orgaan falen (MOF), acuut ischemisch cerebrovasculair accident (CVA) en een cholestatische levercrise
Preoperatieve voorbereiding met top-up of erytroferese/wisseltransfusie wordt nader beschreven in het hoofdstuk Perioperatieve zorg.
|
Indicaties acute transfusie |
Type transfusie |
Score |
|
Aplastische crise |
Top-up transfusie |
A3 |
|
Hb <3,5 mmol/L |
Top-up transfusie |
A3 |
|
Symptomatische anemie |
Top-up transfusie |
A3 |
|
Acuut chest syndroom |
Afhankelijk van ernst, zie hoofdstuk Acuut chest syndroom |
A3 |
|
Miltsequestratie |
Top-up transfusie |
A3 |
|
Acute cholestatische levercrise |
Erytroferese/wisseltransfusie |
A3 |
|
Acuut CVA/TIA |
Erytroferese/wisseltransfusie |
A2 |
|
Multi-orgaan falen |
Erytroferese/wisseltransfusie |
A3 |
|
Preoperatief |
Top-up transfusie of erytroferese/wisseltransfusie, zie hoofdstuk Perioperatieve zorg |
A2 |
|
Hb < 4.5 mmol/L in de zwangerschap en <5,0 mmol/L vlak voor de partus |
Top-up transfusie |
A3 |
B. Chronische erytroferese/wisseltransfusie met een streef HbS<30% voor de volgende wisseltransfusie wordt toegepast ter voorkoming van een (recidief) CVA/TIA na een doorgemaakt CVA of een afwijkende TCD én vasculopathie bij MRA (zie hoofdstuk ischemisch CVA). Indien erytroferese/wisseltransfusie niet haalbaar is, bijvoorbeeld in verband met moeilijke veneuze toegang, kunnen ook top-up transfusies gegeven worden met een streef HbS <30% (dit streefniveau is alleen mogelijk na herhaalde top up transfusies). In uitzonderlijke gevallen kan overwogen worden om bij patiënten met zeer frequente VOCs of andere ernstige complicaties die niet reageren op hydroxyurea, een chronisch erytroferese/wisseltransfusie programma te starten. Dit dient altijd te gebeuren in overleg met een expertisecentrum.
|
Indicaties voor chronische erytroferese/wisseltransfusie |
Score |
|
Primaire preventie van ischemisch CVA (zie hoofdstuk ischemisch CVA) |
A1
|
|
Secundaire preventie ischemisch CVA (zie hoofdstuk ischemisch CVA) |
A2 |
|
Zeer frequente VOC, niet reagerend op hydroxyurea |
C3 |
C. FMS-richtlijn (2019) adviseert dat sikkelcelpatiënten ABO-Rhesus volledig typering, K, Fy(a), en indien mogelijk ook Jk(a), Jk(b) en S compatibel bloed dienen te ontvangen ter preventie van allo-immunisatie. In geval van antistoffen dient altijd rekening te worden gehouden met de gevormde antistof. Zie Startpagina – Bloedtransfusiebeleid.
|
Aanbeveling |
Score |
|
Bij diagnose uitgebreide bloedgroep typering: ABO, Rhesus volledig uittyperen, K, Fy(a), Jk(a), Jk(b) en MNSs |
A3 |
|
Bij erytrocyten transfusie: ABO, Rhesus volledig uittyperen, K, Fy(a), en zo mogelijk ook Jk(a), Jk(b) en S compatibel bloed geven NB: Jk(a), Jk(b) en S in volgorde van belang. |
A3 |
|
Genotypering wordt aangeraden |
C1 |
D. Parvo B19 veilig getest bloed wordt geadviseerd bij alle patiënten met sikkelcelziekte die Parvo B19 IgG negatief zijn.
|
Aanbeveling |
Score |
|
Parvo veilig bloed indien Parvo B19 IgG serologie negatief |
A3 |
Achtergrondinformatie (onderbouwing)
Bij sikkelcelziekte is anemie op zichzelf geen indicatie voor transfusie. Ondanks het ontbreken van bewijs, wordt in de klinische praktijk een absolute ondergrens gehanteerd van 3,5 mmol/L. Bij ernstige klinische situaties (waarbij vooral HbS reductie gewenst is) verdient erytroferese/wisseltransfusie de voorkeur. Een eind Hb > 6.5 mmol/l na transfusie dient vermeden te worden om hyperviscositeit te voorkomen. Zowel voor acute als chronische transfusies wordt een HbS nagestreefd van <30%. In stabiele situaties kan bij volwassen patiënten met secundaire preventie van een CVA een streef HbS% van <50% worden aangehouden. Voor preoperatieve voorbereiding wordt verwezen naar het hoofdstuk Perioperatieve zorg. Bij HbSC patiënten dient het HbS opgeteld te worden bij het HbC (dus HbS + HbC < 30%). Het aantal benodigde erytrocytenconcentraten voor erytroferese/wisseltransfusie is voor volwassenen te berekenen met het volgende algoritme:
rekenblad_erythrocytaferese.xls
Voor kinderen geldt een ander volume en kan het via de volgende tabel:
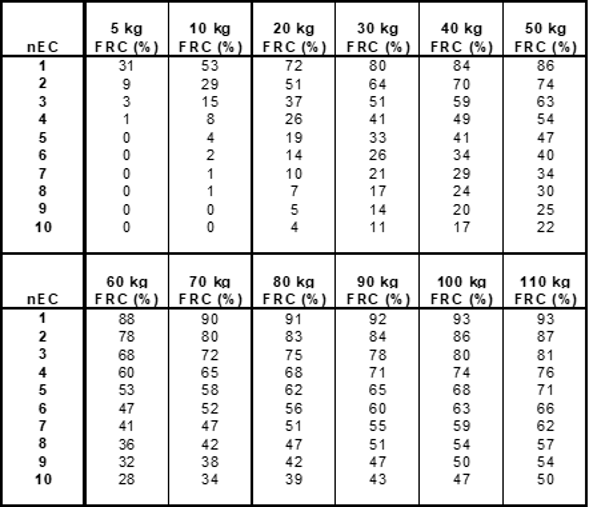
Of via de volgende link: Sickle Cell Rbc Exchange Volume | Medical Calculator (pediatriconcall.com)
Voor de onderbouwing van transfusie indicaties bij sikkelcelziekte-gerelateerde complicaties zoals in bovenstaande tabellen genoemd, verwijzen we naar de desbetreffende hoofdstukken. Bij patiënten met zeer frequente VOC’s zonder respons op hydroxyurea wordt in uitzonderlijke gevallen besloten tot een chronisch transfusie/erytroferese/wisseltransfusie beleid. Alhoewel hier geen gerandomiseerde studies naar gedaan zijn is er voldoende bewijs dat chronisch transfusiebeleid leidt tot minder crisen en acuut chest syndroom.
Bloedtransfusie zonder uitgebreide matching voor Rhesus (CDE) en Kell leidt tot allo-immunisatie in 35% van de patiënten afhankelijk van het aantal transfusies. Een observationele studie liet zien dat extended matching leidt tot minder allo-immunisatie. In de studies waarin niet gematcht wordt, worden naast Rh en K-alloantistoffen, ook antistoffen tegen andere klinisch relevante bloedgroep antigeensystemen gemaakt. Theoretisch leidt uitgebreider matchen tot grote reductie in de vorming van alloantistoffen. Opvallend genoeg zijn de meeste alloantistoffen bij sikkelcelpatiënten tegen Rhesus antigenen gericht. Dit kan worden verklaard door Rhesus varianten die juist bij patiënten oorspronkelijk afkomstig uit Afrika frequenter worden gezien. Matchen voor S is in verband met de geringe beschikbaarheid van eenheden voor preventief matchen vaak niet mogelijk en wordt derhalve ook niet als standaard geadviseerd.
Parvo B19 virusinfecties kunnen bij patiënten met een hemolytische anemie, waaronder patiënten met sikkelcelziekte, leiden tot een passegère aplasie van de erytroïde voorlopercellen. Om die reden wordt geadviseerd Parvo B19 veilig bloed te geven indien een patiënt geen Parvo B19 infectie heeft doorgemaakt (negatieve IgG Parvo B19 serologie). Er zijn geen prospectieve studies die dit beleid onderbouwen.
Bloedtransfusies kunnen gepaard gaan met complicaties, waaronder allo-immunisatie, acute en vertraagde hemolytische transfusiereactie, ijzerstapeling, overdracht van infecties, decompensatio cordis en stijging van de viscositeit van het bloed. Om die redenen dienen bloedtransfusies weloverwogen toegediend te worden.
Zoekverantwoording
Voor de zoekverantwoording van transfusie indicatie bij specifieke sikkelcelziekte gerelateerde indicaties verwijzen we naar de desbetreffende hoofdstukken. Ten aanzien van de allo-immunisatie, is er geen systematische literatuur analyse mogelijk. Voor verdere onderbouwing verwijzen we naar de CBO richtlijn voor bloedtransfusie (2011).
Uitgangsvraag
Aanbeveling
Transfusie gerelateerde hemolyse is het gevolg van allo-antistoffen. Bij een extreme vorm van transfusie gerelateerde hemolyse is het Hb gehalte na transfusie lager dan ervoor (hyperhemolyse).
|
Aanbeveling |
GRADE |
|
A. Indien binnen 28d na transfusie toename van hemolyse optreedt met of zonder VOE-achtige klachten of dyspnoe, dient gedacht te worden aan VHTR. Bepaal aan de hand van het HbA% en met behulp van figuur 1 de waarschijnlijkheid van de diagnose VHTR. Om die reden wordt geadviseerd na elke episodische transfusie < 24 uur een Hb-gehalte en HbA% te bepalen. |
C1 |
|
B. Bij dwingende transfusie indicatie dient antilichaam compatibel bloed toegediend te worden. |
C1 |
|
C. Bij een VHTR met hyperhemolyse dienen erytrocytentransfusies vermeden te worden. Start met prednison en immunoglobulines. Indien dit onvoldoende effect heeft overweeg erytropoëtine, eculizumab en evt combineren met Rituximab. |
D1 |
|
D. Het risico op een VHTR kan worden ingeschat door gebruik van een predictiescore (zie tabel onder). |
C1
|
|
E. Bij patiënten met een verhoogd risico op VHTR dient immunosuppressieve therapie (IVIG, steroiden en/of rituximab) voorafgaand aan de transfusie te worden overwogen. |
D1 |
Onderbouwing
In een prospectieve studie in patiënten met sikkelcelziekte na episodische transfusie werd een incidentie van VHTR van 4,2% gezien. Dit is waarschijnlijk een onderschatting omdat VHTR vaak niet of pas laat gediagnosticeerd wordt en omdat de presenterende klachten kunnen lijken op die van een VOE en/of acuut chest syndroom. Slechts bij een deel van de patiënten zijn antistoffen aantoonbaar, vaak een combinatie van zowel auto- als allo-antistoffen.
Een hyperhemolytische transfusiereactie wordt gedefinieerd als een toename van hemolyse na transfusie met een eind Hb-gehalte lager dan voor transfusie. Een VHTR is de meest voorkomende vorm van transfusiereacties bij patiënten met sikkelcelziekte. Een VHTR wordt zelden gezien bij patiënten met een chronisch transfusieschema.
VHTR wordt meestal geluxeerd door eerder gevormde allo-antistoffen, niet gedetecteerd in de type en screening voorafgaand aan de transfusie omdat de titers onder de detectiegrens zijn gedaald.
Een VHTR word gedefinieerd als significante daling van het Hb-gehalte binnen 28 dagen na transfusie in combinatie met tenminste 1 van de volgende items: nieuw gevormde allo-antistof, hemoglobinurie, relatieve daling van HbA post-transfusie, relatieve reticulocytopenie, significante stijging van LDH en geen andere oorzaak.
Om de waarschijnlijkheid op VHTR te kunnen bepalen wordt het verschil in HbA% tussen het HbA% binnen 24u na transfusie en het HbA% op het moment van klachten. Zie figuur 1 en http://www.reamondor.aphp.fr/nomogram-2/. Een intermediair of hoge waarschijnlijkheid wordt beschouwd als VHTR.
Er dient aandacht te zijn voor optimale respiratoire ondersteuning en oxygenatie. Adequate hydratie en vochtbalans dienen te worden nagestreefd om hemolyse-geïnduceerde tubulopathie en daarmee gepaard gaande acute nierinsufficiëntie te voorkomen. Voorzichtigheid met extra vochttoediening is geboden, vanwege het risico op een verdere daling van het hemoglobinegehalte. Overweeg profylactische antistolling en gebruik van erytropoëtine. Verder is het advies zowel ijzer- als foliumzuursuppletie sterk te overwegen.
Er is casuïstische literatuur, die succesvolle behandeling van transfusie gerelateerde hemolyse met immuunglobulines (IVIG), steroïden, en erytropoëtine laat zien. In een recente “How I treat DHTR” in Blood 2018 is voor zowel de diagnostiek als de behandeling een helder stappenplan omschreven waarbij voor de behandeling van DHTR gestart wordt met:
Er is een predictiescore gedefinieerd (zie tabel 1). Patiënten kunnen op basis van deze score ingedeeld worden in drie risicogroepen: laag risico <8 punten, intermediair risico 8-14 punten, hoog risico >14 punten. Uit dit onderzoek bleek dat vooral laag risicopatiënten goed te definiëren zijn met deze score (negatief-voorspellende waarde 98%, positief-voorspellende waarde 50%). Bij intermediair en hoog risico dient men zeer terughoudend te zijn met erytrocytentransfusies. Bij een strikte indicatie voor een erytrocytentransfusie kunnen immunosuppressieve middelen (IVIG, steroïden en/of rituximab) overwogen worden. Hiervoor is echter geen wetenschappelijk bewijs.
Tabel 1. Risico-inventarisatie voor het inschatten VHTR risico
|
Parameter |
Punten |
|
Voorgeschiedenis VHTR: |
|
|
Ja |
5 |
|
Nee |
0 |
|
Eerdere allo-immunisatie |
|
|
Afwezig |
0 |
|
Alleen Rh/K-antistoffen en/of antistoffen tegen laagfrequente antigenen, autoantistoffen of antistoffen met onbekende specificiteit) |
5 |
|
Aanwezigheid van minstens één significante alloantistof |
6 |
|
Eerder aantal toegediende erytrocytenconcentraten |
|
|
≥12 |
0 |
|
<12 |
8 |
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen.
Uitgangsvraag
Aanbevelingen
A.
In ieder geval is er een harde indicatie voor hydroxyurea voor de volgende patiënten:
B.
|
Aanbevelingen |
Score |
|
Kinderen (vanaf 9 maanden) met HbSS/HbSβ0onafhankelijk van klinische presentatie na geïnformeerde besluitvorming |
A1 (voor kinderen van 9-42 maanden) A2 vanaf 42 maanden oud |
|
HbSS/HbSβ0 patiënten met ≥3 vaso-occlusieve pijncrises per jaar |
A1 |
|
HbSS/HbSβ0 patiënten met sikkelcelziekte-gerelateerd pijn die interfereert met dagelijkse activiteiten en kwaliteit van leven |
A1 |
|
HbSS/HbSβ0 patiënten met status na ernstig (waarvoor beademing noodzakelijk was) of recidiverend acute chest syndroom |
A2 |
|
HbSS/HbSβ0 patiënten met ernstige symptomatische anemie die interfereert met dagelijkse activiteiten en kwaliteit van leven |
A2 |
|
HbSS/HbSβ0 patiënten met chronische nierinsufficiëntie en erytropoëtine gebruik |
C3 |
|
Bij patiënten met andere vormen van sikkelcelziekte kan bij bovenstaande indicaties hydroxyurea overwogen worden in overleg met een expertisecentrum |
B3 |
|
Aanbevelingen |
Score |
|
Start dosering voor volwassen patiënten met HbSS/HbSβ0 15 mg/kg/dag (afronden op 500 mg) en ophogen (500 mg a 8 weken) opgeleide van kliniek en laboratoriumonderzoek (bloedbeeld, lever- en nierfunctie) met een streefdosering van 25 mg/kg. Bij chronische nierinsufficiëntie 5-10 mg/kg/dg |
A1 |
|
Startdosering voor kinderen met HbSS/HbSβ020 mg/kg/dag en ophogen met 5 mg/kg/dg a 8 weken tot maximaal 30-35 mg/kg/dg op geleide van laboratoriumonderzoek (bloedbeeld, lever en nierfunctie |
A1 |
|
Dosisreductie als neutrofielen en/of trombocyten < 1,25 x 109/L of <80 x 109/L zijn |
A3 |
Achtergrondinformatie (onderbouwing)
Hydroxyurea is het enige geneesmiddel waarvan aangetoond is dat het VOC’s kan verminderen. In verschillende observationele en gerandomiseerde studies is aangetoond dat het gebruik van hydroxyurea het aantal VOC’s en de incidentie van acuut chest syndroom grofweg halveert. Tevens leidt het gebruik van hydroxyurea tot een verhoging van het Hb gehalte en afname van de hemolyse. Het voorkomen van orgaanschade door hydroxyurea is niet overtuigend aangetoond. In enkele observationele studies werd wel een verbeterde overleving gezien bij patiënten die hydroxyurea gebruikten. Er waren in deze studies geen aanwijzingen dat eenmaal ontstane eindorgaanschade door gebruik van hydroxyurea ongedaan gemaakt kan worden. Voor kinderen met HbSS/HbSβ0 is het standard beleid dat vanaf 9 maanden hydroxyurea gestart kan worden in overleg met ouders. Voor volwassen patiënten met HbSS/HbSβ0 is het advies ook alle patiënten hydroxyurea aan te bieden maar in ieder geval voor patiënten met frequente crisen ook als daarvoor geen opname noodzakelijk is.
Het werkingsmechanisme van hydroxyurea berust op het verhogen van het foetaal Hb (HbF), maar andere effecten bestaan uit verlaging van het aantal neutrofiele granulocyten, verminderde adhesie van bloedcellen aan de vaatwand, verbeterde NO-beschikbaarheid en afname van de hemolyse.
Hydroxyurea is voornamelijk onderzocht in patiënten met ernstige vormen met sikkelcelziekte (HbSS en HbSβo). In patiënten met andere vormen van sikkelcelziekte zoals HbSC en HbSβ+ zijn nauwelijks studies verricht, en is de effectiviteit niet bewezen. Desondanks is het advies hydroxyurea ook bij deze patiënten te starten voor bovengenoemde indicaties. In alle gevallen dient rekening te worden gehouden met toxiciteit van hydroxyurea. Belangrijke bijwerkingen zijn leukopenie, ulcera aan de enkels, koorts en longinfiltraten. Tot op heden zijn er geen aanwijzingen dat langdurig hydroxyurea gebruik tot een verhoogde kans op leukemie leidt. Wel is het gebruik van hydroxyurea geassocieerd met een hogere kans op plaveiselcarcinoom van de huid. Van hydroxyurea zijn teratogene effecten beschreven en het is gecontra-indiceerd bij zwangere vrouwen of premenopauzale vrouwen zonder effectieve anticonceptie. Hydroxyurea leidt bij mannen tot een verminderde spermatogenese en dient 3 maanden voor de beoogde conceptie gestaakt te worden. Mannen met sikkelcelziekte en oligospermie hebben een kleine kans om na het staken van hydroxyurea onvruchtbaar te blijven. Semen preservatie kan overwogen worden voor start van hydroxyurea. Bewijs voor dit beleid ontbreekt.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen: NHLBI richtlijnen.
Uitgangsvragen
Aanbevelingen
|
Aanbevelingen |
Score |
|
A. Bij een laag risico ingreep: geen transfusie. Patiënten met een laag risico ingreep en algehele anesthesie worden beschouwd als intermediair risico. Bij intermediair risico dient patiënt een top-up transfusie te krijgen met een streef Hb gehalte van 6,5 mmol/L. Patiënten met een preoperatief Hb gehalte hoger dan 5.6 mmol/L, adviseren we een erytroferese/wisseltransfusie met een streef HbS<60% (voor patiënten met HbSC geldt dat HbS + HbC samen <60% moet zijn na erytroferese/wisseltransfusie). Bij een hoog risico ingreep dient altijd erytroferese/wisseltransfusie preoperatief verricht te worden (streef HbS<30%). |
B3
A2
A3 |
|
B. Er dient gestreefd te worden naar normothermie. Een goede hydratietoestand dient gewaarborgd te zijn (3 liter per 24 uur). |
A3 |
|
C. Er dient postoperatief gestreefd te worden naar adequate pijnbestrijding, goede hydratie ((kinderen 2 liter/m2 en volwassenen 3 liter per 24 uur) en tromboseprofylaxe |
A3 |
Achtergrondinformatie (onderbouwing)
Chirurgische ingrepen zijn geassocieerd met het optreden van complicaties bij patiënten met sikkelcelziekte. In een grote observationele studie traden sikkelcelziekte-gerelateerde complicaties zoals VOC, acuut chest syndroom en CVA in 0-18,6% van de patiënten met homozygote sikkelcelanemie op afhankelijk van de ingreep. Niet sikkelcelziekte-gerelateerde complicaties zoals koorts, infectie, trombose, bloeding en dood kwamen in 5,7-26,2% van de patiënten met homozygote sikkelcelanemie voor. In een recente gerandomiseerde studie werd de effectiviteit van preoperatieve bloedtransfusies ter preventie van sikkelcelziekte-gerelateerde complicaties aangetoond. Negenendertig % (13/33) van de patiënten zonder preoperatieve transfusie ontwikkelden een klinisch belangrijke complicatie tegenover 15% (5/34) in de getransfundeerde groep. Acuut chest syndroom werd in geen van de getransfundeerde patiënten gezien en in 9 /33 niet getransfundeerde patiënten. Ten aanzien van het gebruik van een tourniquet is recent een review verschenen. In dit review waarin enkele case series worden beschreven, treedt bij de minderheid van de patiënten een complicatie op. Een individuele afweging ten aanzien van het gebruik van een tourniquet wordt om die reden geadviseerd.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen: NHLBI richtlijnen.
Uitgangsvraag
Wat zijn de adviezen met betrekking tot infectiepreventie middels vaccinaties en antibiotica?
Aanbevelingen
Patiënten met sikkelcelziekte hebben een indicatie voor vaccinatie tegen pneumokokken, Haemophilus Influenzae type b, meningokokken groep B en ACWY en influenza.
|
Vaccinatie |
Schema volwassenen |
|
PCV13 |
1-maal (eenmalig inhalen indien nooit gevaccineerd) |
|
PPV23 |
1 maal (min. 2 maanden na PCV13) |
|
Hib |
1-maal (eenmalig inhalen indien nooit gevaccineerd) |
|
MenACWY |
1-maal |
|
MenB |
1+1 |
|
Influenza |
Jaarlijks |
|
Vaccinatie |
Schema kinderen** |
|
PCV13 |
1-maal rond leeftijd 18-20 maanden |
|
PPV23 |
1 maal (min. 2 maanden na PCV13) op leeftijd 24 maanden/ bij diagnose. Dan op leeftijd 5 jaar. Daarna elke 5 jaar herhalen |
|
Hib |
Via RVP en 1-maal een booster op leeftijd 3 jaar |
|
MenACWY |
Via RVP op leeftijd 14 maanden en booster op leeftijd 4,5-6,5 jr Alle patiënten die sinds 2018 via een inhaalcampagne zijn gevaccineerd met MenACWY, dan wel via RVP bij 14 jaar zijn gevaccineerd, een booster aanbieden na 3-5 jr. |
|
MenB |
MenB vaccinatie aanbieden vanaf leeftijd 2 jr: 1+1 schema met tenminste 1 maand interval. |
|
Influenza |
Jaarlijks vanaf leeftijd 2 jaar (optioneel) |
** wijkt af van LCI richtlijn, want aangepast door de sectie kinderhematologie in 2020
Patiënten met sikkelcelziekte hebben een indicatie voor penicilline profylaxe vanaf de leeftijd van 4 maanden t/m het 5e jaar. Overweeg indien on demand gebruik niet haalbaar is bij kinderen van 6 jaar en ouder, de behandelduur te verlengen tot en met het 12e jaar. Indien het kind ooit een sepsis/meningitis met pneumokok heeft doorgemaakt is er een indicatie om de profylaxe (levens)lang te continueren. Vanaf 6 jaar is het advies over te gaan op een on demand beleid met amoxicilline/clavulaanzuur (Augmentin) bij een temperatuur >38.5˚C. Bij overgevoeligheid voor penicilline wordt claritromycine geadviseerd.
Patiënten met sikkelcelziekte moeten malariaprofylaxe gebruiken bij reizen naar een endemisch gebied
|
Medicatie |
Leeftijd |
Dosering |
|
Feneticilline (Broxil) |
< 1 jaar 1 – 5 jaar 5 – 6 jaar > 6 jaar |
15 mg/kg/dag in 2x (of 2dd 75 mg) 2dd 125 mg 2dd 250 mg op indicatie |
|
Amoxicilline/clavulaanzuur |
Kind Volwassene |
50/12.5 mg/kg/dag in 3 doses 3dd 625 mg |
|
Bij penicilline allergie Claritromycine
Azitromycine |
Kind
Volwassene Kind
Volwassene |
1dd 7.5 mg/kg/dag (profylaxe) 15 mg/kg/dag (therapeutisch) 1dd 500 mg 3x/week 10 mg/kg (profylaxe) 1dd 10 mg/kg/dag (max 500 mg) 1dd 500 mg |
|
Aanbevelingen |
Score |
|
Patiënten met sikkelcelziekte hebben een indicatie voor penicilline profylaxe vanaf de leeftijd van 4 maanden t/m het 5e jaar. |
A2 (geldt voor leeftijd tot 6 jaar) |
|
Patiënten met sikkelcelziekte hebben een indicatie voor vaccinatie tegen pneumokokken (prevenar/pneumovax), Haemophilus Influenzae type b en meningococcen groep ACWY. |
A2 (voor pneumokokken) B3 (voor overige vaccinaties) |
|
Patiënten met sikkelcelziekte moeten malariaprofylaxe gebruiken bij reizen naar endemisch gebied. |
B3 |
Achtergrondinformatie (onderbouwing)
Met name patiënten met HbSS of HbSβ0 thalassemie hebben vanaf de leeftijd van 6 maanden geen functionerende milt meer. Vóór de introductie van profylactische penicilline was sepsis de belangrijkste doodsoorzaak bij kinderen jonger dan 3 jaar, met een mortaliteit van 10%. Een gerandomiseerde studie liet zien dat penicilline profylaxe 84% reductie gaf van infecties. Bij patiënten met een samengesteld heterozygote vorm van sikkelcelziekte zoals HbSC of HbSβ+ thalassemie, is het risico op infecties onduidelijk. Verschillende studies laten zien dat het gevaar op infectieziekten op de jonge leeftijd niet verhoogd lijkt maar er wordt toch geadviseerd hetzelfde beleid toe te passen omdat functionele asplenie ook bij deze patiënten kan ontstaan en aanleiding kan geven tot fatale infecties.
Voor alle patiënten met sikkelcelziekte wordt naast het Rijksvaccinatieprogramma op de kinderleeftijd (DKTP-Hib-HepB, pneumokokken (conjugaat), BMR en Meningokokken ACWY) aanvullende vaccinatie met een 23 valent ongeconjugeerd pneumokokkenvaccin geadviseerd, evenals een jaarlijkse influenza vaccinatie. De vaccinatie met het 23 valente polysaccharide pneumokokken vaccin start op de leeftijd van 2 jaar en dient de eerste keer op 5-jarige leeftijd herhaald te worden en daarna iedere 5 jaar. (zie Vaccinatieschema’s in de richtlijn uit 2018 voor preventie van infecties bij mensen met (functionele) hypo- en asplenie op https://lci.rivm.nl/richtlijnen/asplenie
Dit schema werd in 2020 door de sectie kinderhematologie aangepast omdat deze bij de sikkelcelpatiënten praktisch niet uitvoerbaar is (m.n. te veel vaccinaties op jonge leeftijd toe te dienen). Bij bovenstaand aangepast vaccinatieschema (**) wordt er wel vanuit gegaan dat ouders hun kind laten vaccineren conform rijksvaccinatieprogramma (RVP) en dat ouders en kind compliant zijn m.b.t. de inname van de antibioticaprofylaxe.
1.Pneumokokken vaccinatie (ingevoerd in RVP per 2006):
2. Hib (ingevoerd in RVP per 1993):
3. MenACWY (ingevoerd in RVP per 01-05-2018 voor kinderen 14 maanden + 14 jaar):
4. MenB (niet in RVP):
5. Griepvaccinatie:
Voor patiënten die niet volledig volgens het Rijksvaccinatieprogramma zijn gevaccineerd, wordt een inhaalschema geadviseerd. Zie hiervoor de richtlijn “inhaalschema’s” van het RIVM: https://rijksvaccinatieprogramma.nl/inhaalschema
De effectiviteit van pneumokokken vaccinatie werd reeds jaren geleden aangetoond in twee gerandomiseerde studies die een afname van 90,8% lieten zien van invasieve pneumokokken infecties. In kinderen onder de 5 jaar betrof de afname 93,4%. Ook recentere literatuur bevestigd de voordelen van pneumokokken vaccinatie bij patiënten met sikkelcelziekte.
Hoewel niet aangetoond middels gerandomiseerde studies suggereert de enorme afname van infecties met H. influenza bij patiënten met sikkelcelziekte in landen met een hoog inkomen en reguliere Hib vaccinaties, dat dit vaccin wel degelijk van meerwaarde is.
Hetzelfde geldt voor de meningokok, met name serotype C. In 2018 werd vaccinatie tegen serotype C vervangen door de serotypes ACWY. Recent wordt ook vaccinatie tegen meningokokken B geadviseerd. Omdat de incidentie van invasieve meningokokkenziekte veroorzaakt door meningokokken B laag is, is vóór registratie van het vaccin geen klinische effectiviteitsstudie in Nederland gedaan.
Een recenter systematisch overzicht concludeert dat malariaprofylaxe als zinvol wordt beschouwd.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen.
Uitgangsvraag
Aanbeveling
|
Aanbevelingen |
Score |
|
A. De diagnose acuut chest syndroom wordt gesteld op basis van een nieuw pulmonaal infiltraat op de thoraxfoto met klinische symptomen |
C3 |
|
B. Indien sprake is van een acuut chest syndroom wordt geadviseerd zuurstof te geven, breedspectrum antibiotica te starten, adequate pijnstilling te geven, hydratie te beperken, en erytroferese/wisseltransfusie of top up erytrocyten transfusie indien sprake is van snel toenemende zuurstof behoefte en/of dyspneu. |
A3 |
|
C. Indien er sprake is geweest van een ernstig acuut chest syndroom of recidief, wordt hydroxyurea aanbevolen. |
A2 |
|
D. Allogene stamceltransplantatie zou voor een individuele patiënt overwogen kunnen worden |
C3 |
Achtergrondinformatie (onderbouwing)
Acuut chest syndroom is de meest voorkomende ernstige complicatie van sikkelcelziekte en kent een hoge mortaliteit. Acuut chest syndroom komt bij patiënten met een ernstige vorm van sikkelcelziekte frequent voor. Tweeëndertig procent maakt gedurende het leven een acuut chest syndroom door. Voor patiënten met HbSC en HbSB+ thalassemie is dit 18%. De presentatie van een acuut chest syndroom is identiek aan een pneumonie en kan ontstaan tijdens of kort na een VOC of na een chirurgische ingreep. De meest voorkomende oorzaak van een acuut chest syndroom is een luchtweginfectie. Dit is de reden om breed spectrum antibiotica te geven bij een acuut chest syndroom. De waarde van toevoeging van een macrolide is omstreden. In een grote gerandomiseerde studie bij patiënten met een community acquired pneumonie werd geen toegevoegde waarde gezien van het toevoegen van een macrolide aan een beta-lactam antibioticum. In een prospectieve analyse van infectieuze verwekkers bij 255 opname van patiënten met een acute chest syndroom werd in geen van de patiënten een atypische verwekker gevonden. Andere oorzaken van acuut chest syndroom zijn hypoventilatie, atelectase en vetembolieën. Vroegtijdige herkenning is belangrijk. Respiratoire insufficiëntie en overlijden kunnen worden voorkomen door vroegtijdige top op transfusie of erytroferese/wisseltransfusie. Dagelijkse controle van lever- en nierfunctie als mede hemolyse parameters inclusief bloedbeeld wordt geadviseerd, daar een zeldzaam voorkomend vet-embolie syndroom met multiorgaanfalen zich kan ontwikkelen. Het gebruik van incentive spirometry ter voorkoming van een acute chest syndroom is onzeker. In een kleine prospectieve studie werd een effect gezien van incentive spirometry bij kinderen met een acute vaso-occlusieve crise en thoracale pijn, bij volwassen onder dezelfde condities werd geen effect aangetoond .
Ter voorkoming van een acuut chest syndroom wordt hydroxyurea geadviseerd. In een gerandomiseerde placebo gecontroleerde studie werd 50% reductie gezien van het voorkomen van acuut chest syndroom in patiënten behandeld met hydroxyurea.
Zoekverantwoording
Er zijn geen gerandomiseerde studies verricht naar de effectiviteit van de behandeling van een acuut chest syndroom. Op basis van de expertise van de LWHB en de bestaande richtlijnen zijn wij tot onze aanbevelingen gekomen. NHLBI richtlijnen.
Uitgangsvraag
Aanbeveling
|
Aanbevelingen |
Score |
|
A. Start adequate hyperhydratie met NaCl 0,65% en vraag de uroloog in consult voor evaluatie en behandeling. |
A3 |
|
B. Ter preventie kan worden overwogen: Hydroxyurea 25 mg/kg. 5-α-reductase remmer (finasteride 5mg/dag, indien effectief afgebouwen tot 1mg/dag of dutasteride 0.5mg/dag wat afgebouwd kan worden tot 0.5mg/week. Wisseltransfusies met een streef HbS% van <30%. |
B3
B3
C3 |
Achtergrondinformatie (onderbouwing)
Ten aanzien van de secundaire preventie van priapisme wordt hydroxyurea geadviseerd als eerste keuze. Bij onvoldoende effectiviteit of toxiciteit/non-compliance kan behandeling met een 5-α-reductase remmers (finasteride of dutasteride) geadviseerd worden op basis van ervaring binnen de landelijke werkgroep hemoglobinopathie behandelaren. Een studie heeft in 55 patiënten met frequente episoden van priapisme (1-45 episoden per maand) een afname laten zien bij het gebruik van finasteride in het voorkomen van priapisme. Dutasteride is een langer werkende 5-alfa-reductaseremmer die overwogen kan worden bij onvoldoende response op finasteride. Tenslotte kan overwogen worden wisseltransfusies te stareten om bij onvoldoende response de frequentie van priapisme aanvallen te verminderen. Er is nu een fase 2 studie lopend die mogelijk laat zien dat ook crizanlizumab mogelijk een effect heeft op de frequentie van priapisme maar dat middel is in Europa van de markt gehaald nar de negatieve fase 3 studie naar de effecten op het voorkomen van VOC’s.
Zoekverantwoording
Uitgangsvraag
Aanbevelingen
|
Aanbeveling |
Score |
|
Consult oogarts iedere 3 jaar voor alle patiënten met sikkelcelziekte vanaf 15 jaar of frequenter als retinopathie is vastgesteld. |
A3 |
|
Bij proliferatieve retinopathie is lasercoagulatie geïndiceerd. |
A2 |
Achtergrondinformatie (onderbouwing)
Er is geen bewijs dat screenen op retinopathie de kans op ernstige oogheelkundige complicaties vermindert. In verschillende observationele studies is het vóórkomen van retinopathie onderzocht bij de verschillende genotypen. De incidentie neemt toe met de leeftijd. In een prospectieve studie uit Jamaica werd op 25-jarige leeftijd in 43% van de patiënten met HbSC en 14% van de patiënten met HbSS proliferatieve retinopathie gevonden. In een retrospectieve studie werd aangetoond dat de gemiddelde leeftijd van ontstaan van retinopathie 12,8 jaar is. Prospectieve klinische studies hebben laten zien dat lasertherapie blindheid en glasvochtbloedingen kunnen voorkomen.
In een recente retrospectieve studie in Nederland werd gezien dat het genotype HbSC en het mannelijk geslacht de belangrijkste risicofactoren zijn op retinopathie en de kans op progressie van retinopathie. Over de waarde van flebotomie bij patiënten met een relatief hoog Hb bij HbSC sikkelcelziekte is onzekerheid door het ontbreken van prospectieve studies met een klinisch eindpunt (zie HbSC sikkelcelziekte). Wel is er een indicatie om vaker te screenen bij HbSC (zie hoofdstuk HbSC sikkelcelziekte).
Zoekverantwoording
Uitgangsvraag
Aanbevelingen
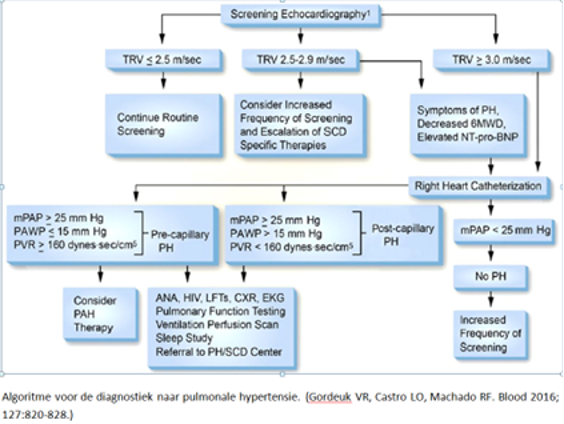
Algoritme voor de diagnostiek naar pulmonale hypertensie
|
Aanbeveling |
Score |
|
A. Verricht voor screening 1x/3 jaar echocardiografie met TRV of sPAP bepaling Bij TRV 2,5 – 2,9 m/s alleen katheterisatie bij klachten of een NT-proBNP > 160 ng/L en anders echo jaarlijks herhalen. Bij TRV ≥ 2,9 m/sec, consulteer een longarts of cardioloog met pulmonale hypertensie expertise voor rechter hartkatheterisatie en overweeg behandeling |
B3
B3
A3 |
|
B. Voor de behandeling van arteriële pulmonale hypertensie (gediagnosticeerd met rechts katheterisatie) is mede behandeling door een in pulmonale hypertensie gespecialiseerde longarts geïndiceerd. Bij post-capillaire pulmonale hypertensie is cardiale evaluatie noodzakelijk. Bij LVEF <50% of globale longitudinale strain (GLS) strain <20% is ook een Holter geïndiceerd gezien het risico op ventriculaire aritmie (zie Cardiomyopathie) |
B2 |
Achtergrondinformatie (onderbouwing)
Screening op pulmonale hypertensie is controversieel. De studie van Desai liet zien dat grofweg 55% van de patiënten met een TRV < of > 2,5 een stijging van de TRV van minimaal 0,1 m/sec in 4,5 jaar hadden. Echter een verhoogde TRV (>2,5 m/sec) is geassocieerd met toegenomen mortaliteit. PH wordt gedefinieerd als een toename van de gemiddelde systolische PAP druk > 25 mmHg gemeten met rechter hartkatheterisatie. PH bij sikkelcelziekte kan het gevolg zijn van pulmonale arteriële hypertensie, pulmonale veneuze hypertensie of PH als gevolg van chronische longziekte of chronische trombo-embolische ziekte. Screening kan plaatsvinden met echocardiografie, door het meten van de tricuspidalis regurgitatie snelheid. In een Franse studie werd aangetoond dat bij slechts 25% van de patiënten met een verhoogde TRV (> 2,5 m/s) werkelijk sprake is van PH bij rechterhartkatheterisatie. (Parent et al.) Een prospectieve studie toonde aan dat verhoogd NTproBNP in combinatie met TRV de positief voorspellende waarde verhoogd (PPV 62%). Er bestaan geen goede placebo-gecontroleerde trials voor de behandeling van PH bij sikkelcelziekte. De beperkte beschikbare literatuur heeft 5 verschillende behandelingen onderzocht, maar deze zijn inconsistent en laten verbetering zien op verschillende eindpunten.
Zoekverantwoording
Voor de diagnostiek is gebruik gemaakt van een systematische review van Musa et al.
Uitgangsvraag
Aanbeveling
|
Aanbeveling |
Score |
|
A. Bij continue of intermitterende pijn wordt een conventionele röntgenfoto geadviseerd, indien niet afwijkend een MRI. |
A3 |
|
B. Behandeling met pijnstilling, fysiotherapie en verwijzing naar orthopedie met specifieke expertise. |
A2 |
Achtergrondinformatie (onderbouwing)
Avasculaire botnecrose wordt bij 20- 50% van de patiënten vastgesteld, waarbij patiënten met HbSS, frequente crisen en/of een relatief hoog Hb, bijv. als gevolg van een bijkomende alfa-thalassemie het hoogste risico hebben. Heupkopnecrose wordt het vaakst beschreven. Het risico neemt toe met de leeftijd. Zowel voor de diagnostiek als voor de behandeladviezen is geen onderbouwing mogelijk daar goede vergelijkende studies ontbreken. Er is 1 kleine gerandomiseerde studie verricht ten aanzien van core-decompression therapie waar geen voordeel voor deze interventie werd aangetoond. De aanbevelingen zijn gebaseerd op expert opinion. Er zijn tegenstrijdige associaties beschreven met betrekking tot hydroxycarbamide gebruik en AVN.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB.
Uitgangsvraag
Wat is het beleid bij asymptomatische en symptomatische cholelithiasis?
Aanbeveling
Er wordt geen electieve cholecystectomie geadviseerd bij patiënten met asymptomatisch galsteenlijden en sikkelcelziekte. Zowel bij volwassenen als bij kinderen met sikkelcelziekte wordt aangeraden een electieve laparoscopische cholecystectomie te doen na een symptomatische episode, zo nodig voorafgegaan door een ERCP. Voor perioperatieve maatregelen verwijzen we naar het hoofdstuk perioperatieve zorg.
Achtergrondinformatie (onderbouwing)
De prevalentie van galstenen neemt toe met de leeftijd van 12% bij 2-4 jarigen, 43% bij 15-18 jarigen en 70-75% op de volwassen leeftijd. Ondanks dit frequente voorkomen treedt acute cholecystitis slechts in 10% van alle patiënten op. Een Jamaicaanse observationele studie in kinderen liet zien dat slechts 2% van de patiënten met galstenen een chirurgische interventie nodig had. De studie van Muroni et al uit 2015 bij volwassen patiënten met SCZ toonde een lager aantal complicaties van profylactische cholecystectomie bij asymptomatische galstenen (4%) versus cholecystectomie na symptomatische cholelithiasis (14%) en ook de opnameduur was iets korter (5.8 vs 7.96 dagen). De review studie van Talhi et al. toont ook een verminderd aantal complicaties van profylactische cholecystectomie bij asymptomatische galstenen bij kinderen.
Er zijn geen gerandomiseerde studies die het voorgestelde beleid (watchful waiting) ondersteunen. Echter bovenstaande resultaten rechtvaardigen volgens de LWHB nog steeds een afwachtend beleid bij asymptomatische patiënten met galstenen.
Zoekverantwoording
Er is geen systematische review of gerandomiseerde studie naar het beleid bij asymptomatisch galsteenlijden, maar bij op basis van bestaande richtlijnen en expertise.
Uitgangsvraag
Aanbeveling
Middels echografie kan intra-hepatische van extra-hepatische cholestase worden onderscheiden. Voor extra-hepatische cholestase verwijzen we naar het hoofdstuk galstenen.
|
Aanbevelingen |
Score |
|
Bij een acute leversequestratie start vochttoediening intraveneus. Corrigeer ernstige Hb daling met bloedtransfusie. |
A3 |
|
Bij een acute intrahepatische cholestase is het advies een sikkelcelexpert te raadplegen voor acute erytroferese/wisseltransfusie. |
A3 |
Achtergrondinformatie (onderbouwing)
Een levercrisis kan onderscheiden worden in acute leversequestratie of acute intra-hepatische cholestase. Acute leversequestratie kenmerkt zich door een acute, pijnlijke leververgroting en een daling in het hemoglobine van > 1,2 mmol/L zonder andere verklaring. Het is een zeldzame complicatie die overwogen dient te worden bij pijn in de rechter bovenbuik. De waarde van aanvullende beeldvorming (bijvoorbeeld CT, MRI, MRCP) naast echografie ter uitsluiting van extra-hepatische oorzaken is niet duidelijk.
Acute intra-hepatische cholestase kenmerkt zich door acute pijn in de rechterbovenbuik, toenemende icterus, vergrote lever en extreme hyperbilirubinemie (zowel ongeconjugeerd als geconjugeerd). De leverenzymstoornissen zijn variabel en er kunnen ook stollingsstoornissen optreden (verlengde aPTT en PT). Het onderscheid met galsteenlijden wordt gemaakt door het uitsluiten van intra- of extra-hepatische galwegobstructie bij stenen. De mortaliteit van patiënten met een ernstige vorm van intra-hepatische cholestase is hoog. In een case-serie wordt een mortaliteit beschreven van 64%. Van deze patiënten overleefden 7 van de 9 patiënten die een erytroferese/wisseltransfusie hadden ondergaan, terwijl slechts 1 van de 13 patiënten zonder erytroferese/wisseltransfusie overleefde. Ondanks dat dit een retrospectieve studie betreft, vormt dit wel de basis van de aanbeveling.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse van de hemoglobinopathie werkgroep en bestaande richtlijnen. Er zijn geen gerandomiseerde studies verricht betreffende levercrise. De aanbevelingen berusten grotendeels op basis van case-reports en case-series.
Uitgangsvraag
Aanbevelingen
|
Aanbevelingen |
Score |
|
A. Het starten met jaarlijkse screening op microalbuminurie wordt aanbevolen vanaf een leeftijd van 4 jaar bij sikkelcelpatiënten met het HbSS of HbSβ0 genotype en bestaan uit hyperfiltratie en micro-albuminurie. |
B2 |
|
B. Bij een microalbumine/kreatinine ratio > 2.5 mg/mmol bij mannen en > 3.5 mg/mmol bij vrouwen dient een ACE inhibitor of een angiotensine receptor blokker gestart te worden. Tevens dient er hydroxyurea gestart te worden, indien patiënt dit nog niet gebruikt. |
B2 |
|
C. Vasculaire risicofactoren inventariseren: controle van bloeddruk (streefbloeddruk 130/80), cholesterolgehalte en nuchtere glucose waarde, nierfunctie en urineonderzoek op micro-albuminurie en proteïnurie. Overweeg erytropoëtine bij (pre-) terminale nierinsufficiëntie, waarbij een maximale bovengrens van het Hb gehalte van 6,2 mmol/l aangehouden dient te worden. Bij terminale nierinsufficiëntie wordt aanbevolen te starten met nierfunctie vervangende therapie en of patiënt door te verwijzen naar een niertransplantatiecentrum. |
B3
B3
A3 |
Achtergrondinformatie (onderbouwing)
De precieze pathofysiologische mechanismen van sikkelcelziekte-gerelateerde nefropathie, het natuurlijk beloop en de optimale behandeling is nog niet opgehelderd. Echter de kennis die opgedaan is bij andere vormen van microalbuminurie en chronische nierfunctiestoornissen is gebruikt in de overwegingen bij de sikkelcelziekte-gerelateerde nefropathie. Aanvankelijk is sikkelcelziekte-gerelateerde nefropathie asymptomatisch waarbij glomerulaire hyperfiltratie (eGFR >200mL/min), glomerulaire hypertrofie en urine concentratiestoornissen (onvermogen te concentreren >450mOsm/kg) reeds in vroege kinderjaren aanwezig zijn. De incidentie en de mate van microalbuminurie (albumine/kreatinine ratio bij herhaling > 2,5 mg/mmol voor mannen en 3.5 mg/mmol voor vrouwen) neemt toe met het stijgen van de leeftijd en leidt uiteindelijk tot progressief nierfunctieverlies. Chronische nierfunctiestoornissen en progressie naar terminale nierinsufficiëntie komt frequent voor bij volwassen sikkelcelpatiënten en gaat gepaard met een aanzienlijke morbiditeit en mortaliteit.
In een retrospectieve observationeel onderzoek met 91 sikkelcelpatiënten (HbSS en HbSβ0) bleek dat de eerste tekenen van hyperfiltratie reeds vanaf 4-jarige leeftijd optrad en dit geleidelijk hierna overging in de aanwezigheid van een progressieve microalbuminurie. Bij ernstige vormen van sikkelcelziekte (HbSS en HbSB0 thalassemie) was in een cohort van volwassen patiënten met een mediane leeftijd van 25 jaar bij 33.9% sprake van microalbuminurie. In een gerandomiseerde studie en enkele observationele studies is aangetoond dat ACE-remming progressie van proteïnurie en microalbuminurie kan voorkomen. In de gerandomiseerde studie werd een afname gezien van de proteïnurie van 37% in de behandelde groep, tegenover een toename van 18% in de placebo behandelde groep na een follow up van een half jaar. Een prospectieve studie door Bartolucci et al. bij patiënten met albuminurie liet zien dat urine albumine/kreatinine ratio significant daalde na 6 maanden hydroxyurea gebruik. Ook bij kinderen en adolescenten is dit effect beschreven.
Het starten met een ACE-remmer of een angiotensine II receptor blokker (ARB) noodzaakt een goede follow-up en controle op mogelijke bijwerkingen. Een bloeddruk van <130/80 mmHg moet worden nagestreefd. De impact van hypertensie voor patiënt gerelateerde uitkomsten is met name significant voor de uit Afrika afkomstige populatie. De screening naar microalbuminurie wordt aanbevolen in een portie ochtendurine of in 2 opeenvolgende porties urine op een willekeurig tijdstip op de dag.
Enkele retrospectieve observationele studies suggereren een gunstig effect van hydroxyurea in de vroege fase van nierinsufficiëntie (hyposthenurie, microalbuminurie en stabilisatie van nierfunctie), en dan met name bij patiënten met een ernstig fenotype (HbSS en HbSβ0) eventueel in combinatie met erytropoëtine. Echter op basis van deze beperkte data kunnen er geen adviezen gegeven worden over het gebruik van hydroxyurea bij de preventie van nierschade. Bij gebruik van erytropoëtine wordt een conservatief maximaal streef Hb gehalte gehanteerd van 6.2 mmol/l, waarboven de dosering moet worden aangepast ter minimalisatie van het risico op vaso-occlusieve complicaties, beroerten en trombotische events.
Er is onvoldoende bewijs dat chronische erytroferese/wisseltransfusie, bloedtransfusie of hydroxycarbamide een gunstig effect heeft op verslechtering van de nierfunctie. De beslissing om een patiënt met terminale nierfunctiestoornissen op een chronische nierfunctie vervangende therapie te laten of door te verwijzen naar een niertransplantatiecentrum moet middels shared-decision making gemaakt worden en is afhankelijk van individuele patiëntfactoren.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse van de hemoglobinopathie werkgroep en bestaande richtlijnen.
Uitgangsvraag
Aanbevelingen
Voor de criteria van pulmonale hypertensie verwijzen we naar het betreffende hoofdstuk.
|
Aanbeveling |
Score |
|
A. Screenen middels echocardiografie en NT-proBNP (jaarlijks) iedere 3 jaar. |
A2 |
|
B. Bij ernstige cardiomyopathie dient het beleid ten aanzien van hartfalen met de cardiologie besproken te worden Bij ernstige anemie (Hb < 5.0 mmol/L) moet chronische transfusietherapie overwogen worden. |
A3 |
Achtergrondinformatie (onderbouwing)
Een studie van Niss et al., toont aan dat screenen van kinderen (mediane leeftijd 10 jaar) bij ongeveer 10% van de patiënten ernstige afwijkingen toont, leidende tot diastolische dysfunctie, wat geassocieerd is met cardiale fibrose. In een recente kleine retrospectieve studie werd gesuggereerd dat de ontwikkeling van cardiale fibrose kan worden beperkt door de vroege introductie van hydroxyurea of chronische transfusie therapie. Er ontbreken goede gerandomiseerde studies welke interventie gekozen moet worden met bij diastolische dysfunctie/cardiale fibrose.
In eerdere studies werd een relatie aangetoond tussen diastolische dysfunctie en mortaliteit. Screenen op cardiale dysfunctie met behulp van echocardiografie wordt geadviseerd, ondanks het ontbreken van studies naar de prospectieve waarde hiervan. De Amerikaanse richtlijn adviseert alleen te screenen bij klachten passend bij pulmonale hypertensie, echter deze klachten zijn aspecifiek en om die reden adviseren wij alle patiënten 3-jaarlijks te screenen op verhoogde pulmonale druk en diastolische dysfunctie. Voor onderbouwing van de behandeladviezen zie paragraaf post-capillaire pulmonale hypertensie.
Recent werd een prospectieve analyse gedaan naar het optreden van ventriculaire aritmie in patiënten sikkelcelziekte waarbij een risicofactor aanwezig was voor hartschade (angina, verhoogd NTproBNP, TRV>2,5, hypertensie, voorgeschiedenis van decompensatio cordis, LVEF<50%, status na allogene stamceltransplantatie, afwijkende ST-segment op een eerder ECG, afwijkende MRI). In deze analyse bleek bij 22% van de patiënten sprake van ventriculaire aritmie bij Holter onderzoek wat bij multivariaat analyse geassocieerd met het mannelijk geslacht, afwijkende GLS bij echocardiografie (maat voor LV-functie) en trombopenie. Om deze wordt bij patiënten met een verhoogd NTproBNP en verminderde linker ventrikelfunctie geadviseerd een cardiologische evaluatie te laten plaats vinden.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse van de hemoglobinopathie werkgroep en bestaande richtlijnen.
Uitgangsvraag
Aanbeveling
|
Aanbevelingen |
Score |
|
A. Bij hypovolemische shock, start direct vochttoediening intraveneus Bloedtransfusie bij ernstige anemie |
A3 A3 |
|
B. Als sprake is van recidiverende miltsequestratie (≥2) of levensbedreigende miltsequestratie wordt splenectomie geadviseerd. |
B3 |
Achtergrondinformatie (onderbouwing)
Acute miltsequestratie wordt gedefinieerd als een acute hemoglobine daling >30% ten opzichte van het normale Hb gehalte van de patiënt en een vergroting van >2 centimeter van de milt ten opzichte van de steady state situatie. Acute miltsequestratie kent een mortaliteit van 15% en is daarmee één van de meest levensbedreigende complicaties van sikkelcelziekte op de kinderleeftijd. De recidiefkans van een levensbedreigende miltsequestratie is 50-80% binnen 4 maanden. Om deze reden is het geïndiceerd om in de stabiele fase direct volgend op de acute behandeling een splenectomie te verrichten. Dit kan in de regel laparoscopisch. Splenectomie bij kinderen met sikkelcelziekte dient in een kinderchirurgisch centrum plaats te vinden. Miltsequestratie kan ook bij volwassen patiënten zich incidenteel voordoen. Het betreft dan meestal patiënten met een compound heterozygote vorm van sikkelcelziekte. Voor hen geldt hetzelfde beleid.
Het advies is niet te veel erytrocyten te transfunderen (maximale Hb 5 mmol/L) omdat de kans op het optreden van hyperviscositeit groot is ten gevolge van een te hoog Hb gehalte na hernieuwd vrijkomen van het in de milt gepoolde bloed. Dit vindt in het algemeen binnen enkele dagen plaats. Men kan zo nodig met behulp van een partiële wisseltransfusie achteraf het Hb gehalte naar beneden corrigeren. Er wordt gestreefd naar een uiteindelijke hematocriet <0.30 l/l.
Zoekverantwoording
Er zijn geen gerandomiseerde studies, de adviezen zijn gebaseerd op observationele studies.
Uitgangsvraag
Aanbeveling
|
Aanbevelingen |
Score |
|
A. Bij 2 separate abnormale TCD (TCDi >185 of TCD>200cm/sec) of bij persisterende (bij herhaling na 1 maand) borderline TCD-waarden (TCDi 170-199cm/sec of TCD 155-179cm/sec) moet een MRA worden verricht.
B. Bij een normale MRA kan overwogen worden van transfusie therapie af te zien, en dient de MRA iedere 2 jaar herhaald te worden bij persisterend verhoogde TCD. Bij vasculopathie moet gestart worden met transfusie therapie wat bij normalisatie van de MRA na 1 jaar of langer omgezet kan worden in hydroxyurea. |
A2
B2 |
|
C. Bij een acuut ischemisch CVA dient een acute erytroferese/wisseltransfusie met een streef HbS <30% te gebeuren. Indien wisseltransfusie <2 uur na presentatie niet haalbaar is wordt top-up transfusie geadviseerd mits Hb <5.5 mmol/L. |
A3 |
|
D. Oudere volwassen patiënt met een acuut ischemisch CVA én cardiovasculaire risicofactoren dient in samenspraak met de neurologie trombolyse overwogen te worden. |
C3 |
|
E. Na het CVA dient een chronische erytroferese/wisseltransfusie programma gecontinueerd te worden met een streef HbS <30%. Allogene stamceltransplantatie moet sterk overwogen worden. Bij het niet haalbaar zijn van HSCT of erytroferese/wisseltransfusie, kan hydroxyurea worden overwogen. |
A3 |
Achtergrondinformatie (onderbouwing)
Kinderen met een verhoogde TCD hebben een sterk verhoogd risico op een ischemisch CVA (10%). In de STOP-trial, een prospectief gerandomiseerde studie, werd aangetoond dat chronische transfusie therapie een 92% reductie van de kans op een ischemisch CVA opgeleverde. Naar aanleiding van deze studie wordt bij alle kinderen vanaf 2 jaar een jaarlijkse TCD geadviseerd en transfusietherapie geïnitieerd indien afwijkend (voor grenswaarden zie boven). In Nederland is de afgelopen 20 jaar een ander beleid gevoerd dan in de meeste richtlijnen staat. Bij afwijkende TCD wordt in Nederland een MRA verricht. Alleen bij middels MRA aangetoond vasculopathie wordt chronische transfusie therapie gestart voor een periode van minimaal 1 jaar. Bij normalisatie van de TCD na 1 jaar kan dit langzaam worden over gezet op hydroxyurea gedoseerd tot de maximaal tolereerbare dosis. Bij patiënten met een normale MRA dienen de TCD-controles 2x/jaar gecontinueerd te worden en de MRA iedere 2 jaar herhaald te worden indien TCD verhoogd blijft. De retrospectieve analyse van dit beleid toonde, in navolging van eerder publicaties uit Frankrijk, dat dit beleid veilig blijkt en de transfusiebehoefte sterk reduceert.
Bij patiënten met sikkelcelziekte die zich presenteren acuut ischemisch CVA wordt acute erytroferese/wisseltransfusie geadviseerd met een streef HbS van 30% in overleg met een expertisecentrum. In 1 retrospectieve studie wordt melding gemaakt van een voordeel van erytroferese/wisseltransfusie versus top up transfusie bij een acuut ischemisch cerebraal event, zo snel mogelijk na het vaststellen van de diagnose5. Indien wisseltransfusie niet binnen 2 uur na presentatie kan worden gedaan wordt geadviseerd aan patiënten met een Hb< 5.5 mmol/L een top-up transfusie te geven tot een Hb van maximaal 6.5 mmol/L.
Na de acute fase van het ischemisch CVA is de kans op een recidief ischemisch CVA tussen de 47-90%. Er is geen vergelijkend onderzoek over de precieze waarde van bloedtransfusies na ischemisch CVA. Op basis van verschillende observationele studies, wordt geadviseerd chronisch transfusietherapie of erytroferese/wisseltransfusie te continueren. Het is onduidelijk wanneer deze preventieve therapie gestaakt kan worden. Hydroxycarbamide ter preventie van een recidief ischemisch CVA lijkt inferieur ten opzichte van transfusietherapie. In de SWITCH-trial was de recidiefkans 10% in de patiënten behandeld met hydroxycarbamide versus 1,5% in de transfusie behandelde groep. Mogelijk wordt dit verschil deels verklaard door de acute switch. In een eerdere studie werd aangetoond dat bij een langzame overgang van transfusietherapie naar hydroxyurea het risico op recidief CVA aanzienlijk lager was. Na de acute fase van het ischemisch CVA is, voornamelijk bij volwassen patiënten, adequate cardiovasculaire risicomanagement geïndiceerd. Er is geen bewijs voor een gunstig effect op overleving of recidiefkans van plaatjesaggregatieremming. Een andere potentiële behandeling waarmee een recidief CVA voorkomen kan worden is een allogene stamceltransplantatie.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse van de hemoglobinopathie werkgroep en bestaande richtlijnen.
Uitgangsvraag
Welke behandeling is het meest effectief bij een patiënt met een ulcus cruris?
Aanbeveling
|
Aanbevelingen |
Score |
|
A. Verwijs naar de afdeling dermatologie bij een ulcus cruris |
A3 |
|
B. Neem wondkweken af bij verdenking op infectie en sluit osteomyelitis uit met behulp van beeldvorming. |
B3 |
Achtergrondinformatie (onderbouwing) 2.5% van de patiënten met homozygote sikkelcelziekte ontwikkelt pijnlijke, chronische ulcera aan de onderbenen tussen het 10e en het 50e levensjaar. Ulceraties zijn ongebruikelijk bij de andere genotypen. Het wordt frequenter bij mannen dan vrouwen waargenomen. Mogelijk worden ulcera veroorzaakt door lokale ischemie en infarcering van de huid. Meestal bevinden de ulcera zich aan de mediale of laterale malleolus, zijn vaak solitair, maar multipele ulceraties zijn niet ongebruikelijk. Zij hebben vaak een hardnekkig, recidiverend karakter en zijn zeer pijnlijk, soms gecompliceerd door secundaire infecties.
Er zijn slechts enkele gerandomiseerde studies die verschillende topicale behandelingen hebben vergeleken. Deze studies zijn echter klein. Bij geïnfecteerde ulcera zijn antibiotica op geleide van de kweken geïndiceerd. In een van de studies werd bij geïnfecteerde ulcera een goed effect gezien van een aerosol solution (neomycine, bacitrin en polymyxin B). Het is belangrijke om lokale compressie te vermijden.
Bij patiënten die behandeld worden met hydroxyurea kan worden overwogen over te stappen op wisseltransfusies. Hydroxyurea gebruik, veneuze en arteriële insufficiëntie en diabetes mellitus vergroten de kans op ulcera. Ondanks het feit dat ulcera ook bij patiënten zonder sikkelcelziekte worden gezien bij het gebruik van hydroxyurea werd in een studie gesuggereerd dat de relatie tussen hydroxyurea en ulcera discutabel is bij patiënten met sikkelcelziekte. Ook is de toegevoegde waarde van bloed-/wisseltransfusie bij patiënten met sikkelcelziekte is niet aangetoond.
Zoekverantwoording
Uitgangsvraag
Aanbeveling
|
Aanbevelingen |
Score |
|
A. Bij secundaire hemochromatose wordt MRI, laboratoriumonderzoek en botscintigrafie geadviseerd |
A3 |
|
B. Start ijzerchelatie als liver iron concentration (LIC, gemeten middels MRI) >3 mg/g drooggewicht is en/of als >20 erytrocyteneenheden zijn gegeven |
A2 |
|
C. Eerste keus deferasirox, tweede keus deferipron en derde keus is deferoxamine |
A2 |
Achtergrondinformatie (onderbouwing)
IJzerstapeling ontstaat ten gevolge van een toegenomen ijzerabsorptie en/of bloedtransfusies. Het lichaam kent geen mechanisme om overtollig ijzer te verwijderen. Een eenheid erytrocytenconcentraat bevat ± 200 mg ijzer (en opzichte van een dagelijkse resorptie van ijzer uit de voeding van 1-2 mg/dag). De monitoring van ijzerstapeling is niet eenvoudig. Het meest gebruikt voor de follow-up is de serum ferritine concentratie en de ijzersaturatie of transferrine saturatie. Beide bepalingen zijn echter gevoelig is voor acute fase fluctuaties. De gouden standaard is daarom nog altijd MRI van de lever. IJzerstapeling van het hart komt in vergelijking met transfusie afhankelijke thalassemie patiënten bij sikkelcelziekte veel minder voor. De exacte reden daarvoor is nog grotendeels onbekend. Sikkelcelpatiënten lijken een 3,5x lager risico te hebben op het ontwikkelen van orgaanschade (met name hartfalen en hypogonadisme) ten opzichte van thalassemie patiënten bij een gelijke ijzerbelasting.
IJzerchelatie wordt gestart op basis van uitslagen van MRI die uitstekend correleren met liver iron concentration (LIC). Over het effect van chelatietherapie op het ontwikkelen van orgaanschade en het effect op overleving bij sikkelcelziekte is weinig bekend. Gezien het evidente bewijs van het nut van chelatietherapie op overleving bij patiënten met thalassemie worden dezelfde doelstellingen nagestreefd bij patiënten met sikkelcelziekte.
Zoekverantwoording
Uitgangsvraag
Welke preconceptionele adviezen gelden voor patiënten met sikkelcelziekte?
Aanbeveling
|
Aanbevelingen |
Score |
|
A. Partnerscreening op hemoglobinopathie en indien drager verwijzen naar klinisch geneticus |
A3 |
|
B. Counseling bij een klinisch geneticus is geïndiceerd als de partner drager is van een hemoglobinopathie of bèta-thalassemie |
A3 |
|
C. Teratogene medicatie indien 3 maanden voor conceptie gestaakt te worden (hydroxyurea, ACE-remmers en chelatietherapie) |
B2 |
|
D. Mannelijke patiënten wordt geadviseerd hydroxyurea minimaal 3 maanden voor conceptie te staken |
C3 |
Achtergrondinformatie (onderbouwing)
Jaarlijks dient aandacht besteed te worden aan een kinderwens. Het belang van partnerscreening en de consequenties voor het kind dienen bij een partner met een hemoglobinopathie (bijv. HbS, HbC, HbD of HbE en dragerschap voor bèta-thalassemie) besproken te worden met een klinisch geneticus. Indien er sprake is van een risico op een kind met een ernstige hemoglobinopathie kan antenatale diagnostiek aangeboden worden en eventueel pre-implantatie genetische diagnostiek, waarbij met behulp van IVF, embryoselectie kan worden toegepast.
Teratogene medicatie, zoals hydroxyurea, ACE-remmers en chelatietherapie dienen minimaal 3 maanden voor de conceptie gestaakt te worden bij de vrouwelijke patiënt. Er zijn echter aanwijzingen dat hydroxyurea veilig kan worden gebruikt tot aan conceptie. Mannelijke patiënten wordt geadviseerd 3 maanden voor conceptie hydroxyurea te staken. Alhoewel er geen literatuur bestaat over de potentiele teratogene effecten van hydroxyurea gebruik bij mannen.
Zoekverantwoording
Uitgangsvragen
Aanbevelingen
|
Aanbevelingen |
Score |
|
A. Zwangere patiënten dienen te worden begeleid in centra met multidisciplinaire zorg en expertise op het gebied van sikkelcelziekte. |
A3 |
|
B. Chronische bloedtransfusie wordt niet standaard geadviseerd in de zwangerschap. Indicaties die bloedtransfusies rechtvaardigen zijn: tweelingzwangerschap, eerdere gecompliceerd verlopen zwangerschap en patiënten met een al bestaande indicatie voor chronische erytrocytaferese. Het streef Hb is dan >4.5 mmol/L. |
B2
|
|
C. Lage-dosis aspirine vermindert het risico op pre-eclampsie onder vrouwen die een verhoogd risico op pre-eclampsie hebben. Postpartum wordt tromboseprofylaxe geadviseerd tot 2wk na vaginale partus. Na een sectio caesarea dient dat tot 6 wkn postpartum voortgezet te worden. Bij hoog risicopatiënten wordt gedurende de gehele zwangerschap tromboseprofylaxe geadviseerd tot 6 weken postpartum in overleg met de vasculaire geneeskunde. |
B3 |
|
D. Een sectio caesarea wordt beschouwd als een intermediair risico ingreep, voor preoperatieve maatregelen zie hoofdstuk perioperatieve zorg. |
A3 |
Achtergrondinformatie (onderbouwing)
Zwangere patiënten met sikkelcelziekte hebben een verhoogde kans op foetale en maternale complicaties. Maternale complicaties zijn met name overlijden, een toename in sikkelcelziekte-gerelateerde complicaties (zoals vaso-occlusieve crisen, acute chest syndroom, infecties) en een toename in zwangerschap-gerelateerde complicaties (zoals pre-eclampsie, zwangerschaps-geïnduceerde hypertensie, trombo-embolieën). Foetale complicaties betreffen spontane abortus, intra-uterien vruchtdood, perinatale sterfte, vroeggeboorte en foetale groeiretardatie
Om die reden dienen zwangere sikkelcelpatiënten in een centrum met expertise op het gebied van sikkelcelziekte te worden begeleid. Gezamenlijke begeleiding door de gynaecoloog en internist-hematoloog wordt sterk aangeraden. Bij patiënten die zwanger geworden zijn tijdens gebruik van hydroxyurea, dienen de mogelijke gevolgen besproken te worden en een uitgebreide prenatale screening aangeboden worden verrichten bij 20 weken. Zie voor het risico van hydroxyurea bij zwangerschap zie hoofdstuk Preconceptioneel advies.
Bij zwangere vrouwen die een hoog risico op pre-eclampsie hadden, is in een meta-analyse gevonden dat gebruik van lage dosis aspirine het risico op het ontwikkelen van pre-eclampsie met 17% wordt verlaagd. Een hoog risico op pre-eclampsie werd hierbij gedefinieerd als de aanwezigheid van hypertensie, aanwijzingen voor een intra-uteriene groeiretardatie, meerlingzwangerschap, voorgeschiedenis van pre-eclampsie, aanwezigheid van nierfunctiestoornis en aanwezigheid van diabetes mellitus. In een overzichtsartikel gebaseerd op deze meta-analyse wordt bij zwangere sikkelcelpatiënten die een hoog risico op pre-eclampsie hebben, aanbevolen te starten met een lage dosis aspirine vanaf 12 weken zwangerschap.
Chronische transfusie bij zwangere sikkelcelpatiënten is een punt van discussie. In een recente meta-analyse was de uitkomst dat transfusies vooral sikkelcel-gerelateerde complicaties kunnen reduceren wat ook uit de enige gerandomiseerde studie van Koshy uit 1988 kwam. Gegeven de forse belasting van zwangerschap voor vrouwen met sikkelcelziekte is er internationaal een neiging patiënten met sikkelcelziekte tijdens de zwangerschap laagdrempeliger te transfunderen. Hier is echter geen consensus over.
Bij zwangere vrouwen wordt laagdrempelig opname geadviseerd en is in ieder geval geïndiceerd als sprake is van koorts, acute Hb-daling of lage zuurstofsaturatie. Tijdens een opname wordt tromboseprofylaxe geadviseerd.
Zoekverantwoording
Uitgangsvraag
Aanbeveling
|
Aanbeveling |
Score |
|
A. Overweeg HSCT bij ongecompliceerde sikkelcelziekte indien HLA-identieke donor beschikbaar is en overweeg in ieder geval HSCT bij kinderen met complicaties ondanks medicamenteuze profylaxe. |
B2 |
|
B. Allogene HSCT kan worden gecompliceerd door mortaliteit (1-5%), rejectie (10%), graft versus host ziekte (2-10%) en infertiliteit. |
B2
|
Onderbouwing
HSCT met gebruik van donor stamcellen is momenteel de enige curatieve behandelmodaliteit voor sikkelcelziekte. Hoe jonger de leeftijd ten tijde van HSCT, hoe beter de uitkomsten. Maar de keuze voor HSCT en het moment van uitvoeren zijn zeer lastig voor kinderen/ouders en behandelaren in verband met HSCT-risico’s. Deze betreffen vooral de toxiciteit van de chemotherapie en het risico op graft-versus-host-ziekte (GVHD). Deze risico’s dienen op individueel niveau zorgvuldig afgewogen te worden tegen de risico’s van sikkelcelziekte gerelateerde (irreversibele) complicaties. In deze richtlijn geven we handvatten die behulpzaam zijn in het maken van deze moeilijke overwegingen voor kinderen 0-18 jaar.
Allogene HSCT, kent ondanks >90-95% disease free survival, complicaties die tot morbiditeit (10-15%) en mortaliteit (1-5%) kunnen leiden. Diverse studies laten betere uitkomsten zien van HSCT gerelateerd aan jongere leeftijd. Dit is deels doordat HSCT minder gecompliceerd verloopt in jonge kinderen vergleken met oudere kinderen en deels door reeds opgetreden irreversibele sikkelcelziekte gerelateerde schade in oudere kinderen. Aan de andere kant, kan jongere HSCT-leeftijd ook nadelen hebben. Zo kan de chemotoxiciteit met infertiliteit tot gevolg een reden zijn om af te zien van HSCT. Mogelijkheid van ovariumcryopreservatie op telkens jongere leeftijd en testisbiopsie bieden hier verbeteringen voor de toekomst.
Bij beschikbaarheid van een HLA-identieke donor is onder experts toenemend consensus om over te gaan tot transplantatie, zelfs bij asymptomatische patiënten op het moment van HSCT. Een zorgvuldige beslissing kan niet gemaakt worden zonder de volgende overwegingen: mate van ziektelast en ziektecomplicaties, wensen van de patiënt en ouders na volledige informatievoorziening (zowel over de ziekte als HSCT), beschikbaarheid donoren en draagkracht.
Het intensieve stamceltransplantatie traject vraagt optimale post-transplantatie zorg met specifieke aandacht voor sikkelcelziekte gerelateerde late effecten, conform de richtlijn “Late effecten follow-up bij kinderen na stamceltransplantatie of celtherapie met benigne hematologische, metabole of immunologische indicaties”.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen met de volgende referenties.
Uitgangsvraag
Aanbeveling
|
Aanbevelingen |
Score |
|
A. alle patiënten met sikkelcelziekte-gerelateerde complicaties ongeacht donor beschikbaarheid. Deze complicaties zijn:
Overweeg bij beschikbaarheid van een HLA identieke broer/zus HSCT bij alle patiënten met ernstige genotypen HbSS of HbS-β0-thalassemie, ongeacht complicaties. |
B2
B2
|
|
B. rejectie, infectieuze complicaties en acute graft versus host ziekte |
B2
|
Achtergrondinformatie (onderbouwing)
Gebruik van niet-verwante donoren wordt vooralsnog niet aanbevolen bij volwassen sikkelcelpatiënten. Informatie voorziening en shared-decision making wordt geadviseerd in een expertise centrum op het gebied van HSCT bij patiënten met sikkelcelziekte.
Verschillende studies hebben aangetoond, dat de kwaliteit van leven verbetert na het ondergaan van een allogene stamceltransplantatie. Er is nog geen direct bewijs dat HSCT het leven verlengt ten opzichte van sikkelcelpatiënten die geen HSCT ondergaan. Studies met een gematchte controlegroep en/of een voldoende lange follow-up duur ontbreken. Het is echter wel aannemelijk dat een succesvolle HSCT de levensverwachting van de getransplanteerde patiënt zal verlengen.
Door de ontwikkeling van niet-myeloablatieve conditioneringsschema’s met alemtuzumab en lage dosis totale lichaamsbestraling (TBI) is het gebruik van HSCT met een broer/zus als donor bij volwassen patiënten de laatste jaren sterk toegenomen. De 5-jaars overleving en ziektevrije overleving na transplantatie met een HLA-identieke sibling donor en alemtuzumab/3Gy TBI waren respectievelijk 93% en 85% in een studie met 122 patiënten uit drie verschillende centra. De toxiciteit na alemtuzumab/TBI conditionering is mild, er treedt praktisch geen graft-versus-host ziekte op en de fertiliteit blijft behouden. Er is daarentegen wel een reële kans op afstoting en dus terugkeer van sikkelcelziekte (10-15%).
Een systematische review naar de uitkomsten van haplo-identieke donortransplantaties voor sikkelcelziekte heeft laten zien, dat deze uitkomsten in de loop van het laatste decennium sterk zijn verbeterd. Naast meerdere myeloablatieve schema’s, zijn er niet-myeloablatieve conditioneringen bestaande uit ATG, lage dosis chemotherapie, lage dosis TBI en PTCy als GvHD profylaxe ontwikkeld. De 1-jaars overleving in de meest recent gepubliceerde onderzoeken bij volwassen patiënten was 87-100% met een graft failure percentage van 7-13%. De toxiciteit na niet-myeloablatieve haplo-identieke transplantaties bij volwassen patiënten met sikkelcelziekte is relatief mild en vooral gerelateerd aan de conditionering (chemotherapie/TBI), het gebruik van immuunsuppressie (sirolimus) en het optreden van GvHD en infecties.
De plaatsbepaling van HSCT ten opzichte van gentherapie is op dit moment nog onduidelijk. Klinische studies naar gentherapie met ex-vivo gemanipuleerde autologe hematopoëtische stamcellen met verschillende strategieën (waaronder inductie van foetaal hemoglobine en correctie van de pathogene mutatie zelf) zijn gaande. Terwijl de eerste resultaten veelbelovend zijn, is een aantal belangrijke vragen ten aanzien van lange termijn complicaties van gentherapie en de toxiciteit van de myeloablatieve conditionering, vooral bij volwassen patiënten, nog onbeantwoord. Gentherapie, al dan niet in studieverband is vooralsnog niet beschikbaar in Nederland.
Ten aanzien van de risico’s ten gevolge van HSCT toonde een grote serie van HSCT met een HLA-identieke sibling donor na niet-myoablatieve conditionering met alemtuzumab/3Gy TBI een mortaliteit van 7% en een rejectie percentage van 15% aan. Zowel acute als chronische GVDH treedt niet tot nauwelijks op. Immuunsuppressie gedurende minimaal 1 jaar met sirolimus leidt wel regelmatig tot botpijnen.
Zoekverantwoording
Er is geen systematische literatuur analyse verricht, maar gerichte analyse op basis van expertise van de LWHB en de bestaande richtlijnen.
Uitgangsvraag
Aanbevelingen
|
Conclusie |
Score |
|
Screening voor retinopathie 1x/jaar wordt sterk aanbevolen bij HbSC ziekte |
B2 |
|
Flebotomie kan worden overwogen bij patiënten met een Hb >7.0 of Ht >0.35 en frequente vaso-occlusieve crises, met een streef Hb van <6.5 mmol/L |
C3 |
|
Hydroxyurea kan worden overwogen bij frequente crisen bij een Hb <7.0 of een Ht <0.35 |
C3 |
Onderbouwing
De pathofysiologie van HbSC ziekte is wezenlijk anders gezien de combinatie van afwijkend hemoglobine S met hemglobine C. Acute complicaties (VOC en acute chest syndroom) komen voor, maar minder frequent vergeleken met andere vormen van sikkelcelziekte. Een acuut CVA wordt in het algemeen pas op latere leeftijd (volwassen leeftijd) gezien en komt ook minder vaak voor bij HbSC ziekte. Veel voorkomende chronische complicaties bij HbSC ziekte zijn retinopathie (44-70%), gehoorschade (30-47%), avasculaire botnecrose (osteonecrose) (12-28%), priapisme (5-20%) en nefropathie (2-13%).
De ernst van de anemie is over het algemeen minder bij HbSC ziekte. Een recente studie liet zien dat de meeste patiënten een hemoglobine gehalte hebben >6.2 mmol/L. Daarnaast is er sprake van een hogere viscositeit, veroorzaakt door een hogere hematocriet. Flebotomie vermindert direct de viscositeit van het bloed doordat het hematocriet wordt verlaagd. Daarnaast wordt door ijzergebrek het MCHC verlaag waardoor de intracellulaire hemoglobine concentratie lager wordt wat zou kunnen leiden tot afname van polymerisatie. Dit is echter alleen in een muizenmodel onderzocht.
Er zijn enkele case-series waarin een positief effect van flebotomie op de frequentie van VOC wordt beschreven in HbSC en ook HbSS sikkelcelpatiënten met een relatief hoog Hb. Ten aanzien van het effect op proliferatieve retinopathie of AVN zijn geen data in de literatuur beschikbaar en kan derhalve geen advies over worden gegeven.
Data over effectiviteit van hydroxyurea als behandeling voor HbSC ziekte ontbreken. Enkele recente studies laten gunstige effecten zien, met vermindering van VOC. Behandeling met hydroxyurea kan echter ook een stijging van het hemoglobine tot gevolg hebben, waardoor het aanbevolen wordt dit alleen in te zetten bij een Hb <7.0 of Ht <0.35.
Zoekverantwoording
Er is gebruikt gemaakt van bestaande richtlijnen en literatuur search voor wat betreft flebotomie als behandelingsoptie in patiënten met HbSC.
Hydroxyurea is een van de belangrijkste pijlers van de behandeling voor patiënten met sikkelcelziekte. Zie hoofdstuk Hydroxyurea.
In de afgelopen jaren zijn nieuwe disease modifying drugs op de markt gekomen in de VS, waarvan sommige in Europa goedgekeurd zijn maar nog niet voorgeschreven kunnen worden in Nederland.
Dit betreft:
L-glutamine
L-glutamine is een aminozuur die nodig is voor onder andere de synthese van glutathion en arginine. In essentie beschermt het tegen oxidatieve stress, maar de exacte werking bij sikkelcelziekte is onduidelijk. In een fase 3 placebo-gecontroleerde gerandomiseerde studie bij patiënten met ernstige sikkelcelziekte werd een 25% reductie gezien in het aantal vaso-occlusieve crises in 48 weken en een reductie van 33% van opnames voor een pijncrise. Tevens werd een significant verschil gevonden in het optreden in acute chest syndroom. De EMA heeft het geneesmiddel niet toegelaten tot de Europese markt o.b.v. het ontbreken van de gegevens over het werkingsmechanisme en onvoldoende gegevens over adolescenten met sikkelcelziekte
Voxelotor
Voxelotor is een oraal beschikbare polymerisatie remmer van HbS wat werkt via verhoging van de zuurstofaffiniteit van hemoglobine S. In een fase 3 placebo-gecontroleerde gerandomiseerde studie bij patiënten met sikkelcelziekte en een Hb lager dan 6,5 mmol/L werd aangetoond dat voxelotor in een dosering van 1500 mg per dag in 51% van de patiënten leidde tot een Hb stijging van minstens 0,6 mmol/L versus 7% in de placebo groep. Dit effect was onafhankelijk van het gebruik van hydroxyurea. Er werd geen significant verschil aangetoond tussen de behandel arm en de placebo arm in biomarkers van hemolyse en incidentie van vaso occlusieve crise. De indicatie voor dit middel lijkt te zijn voor patiënten met een ernstige hemolytische anemie (Hb gehalte van < 5,5 mmol/L) met onvoldoende response op hydroxyurea of intolerantie voor hydroxyurea. Belangrijk is dit middel nooit acuut te staken maar af te bouwen. Het middel is inmiddels goed gekeurd voor de Europese markt maar nog niet vergoed in Nederland.
Zoekverantwoording
Strength-of-Recommendation Taxonomy (SORT)
|
Code |
Definition |
|
A |
Consistent, good-quality patient-oriented evidence * |
|
B |
Inconsistent or limited-quality patient-oriented evidence * |
|
C |
Consensus, disease-oriented evidence *, usual practice, expert opinion, or case series for studies of diagnosis, treatment, prevention, or screening |
* Patient-oriented evidence measures outcomes that matter to patients: morbidity, mortality, symptom improvement, cost reduction, and quality of life. Disease-oriented evidence measures immediate, physiologic, or surrogate end points that may or may not reflect improvements in patient outcomes (e.g. blood pressure, blood chemistry, physiologic function, pathologic findings).
Assessing Quality of Evidence
|
Study quality |
Diagnosis |
Treatment/prevention/ screening |
Prognosis |
|
Level 1: good-quality, patient-oriented evidence |
Validated clinical decision rule SR/meta-analysis of high-quality studies High-quality diagnostic cohort study* |
SR/meta-analysis or RCTs with consistent findings High-quality individual RCT† All-or-none study‡ |
SR/meta-analysis of good-quality cohort studies Prospective cohort study with good follow-up |
|
Level 2: limited-quality |
Unvalidated clinical decision rule SR/meta-analysis of lower quality studies or studies with inconsistent findings Lower quality diagnostic cohort study or diagnostic case-control study |
SR/meta-analysis of lower quality clinical trials or of studies with inconsistent findings Lower quality clinical trial Cohort study Case-control study |
SR/meta-analysis of lower quality cohort studies or with inconsistent results Retrospective cohort study or prospective cohort study with poor follow-up Case-control study Case series |
|
Level 3: other evidence |
Consensus guidelines, extrapolations from bench research, usual practice, opinion, disease-oriented evidence (intermediate or physiologic outcomes only), or case series for studies of diagnosis, treatment, prevention, or screening |
||
*High-quality diagnostic cohort study: cohort design, adequate size, adequate spectrum of patients, blinding, and a consistent, well-defined reference standard.
†High-quality RCT: allocation concealed, blinding if possible, intention-to-treat analysis, adequate statistical power, adequate follow-up (greater than 80 percent).
‡In an all-or-none study, the treatment causes a dramatic change in outcomes, such as antibiotics for meningitis or surgery for appendicitis, which precludes study in a controlled trial.
(SR = systematic review; RCT = randomized controlled trial)
Consistency Across Studies
|
Consistent |
Most studies found similar or at least coherent conclusions (coherence means that differences are explainable). or If high-quality and up-to-date systematic reviews or meta-analyses exist, they support the recommendation. |
|
Inconsistent |
Considerable variation among study findings and lack of coherence or If high-quality and up-to-date systematic reviews or meta-analyses exist, they do not find consistent evidence in favor of the recommendation. |
Algorithm: assigning a Strength-of-Recommendation grade based on a body of evidence
© 2022. Alle rechten voorbehouden
Nederlandse Vereniging voor Hematologie (NVvH)
Uiterlijk in 2022 bepaalt de LWHB of deze richtlijn nog actueel is. Zo nodig wordt een nieuwe werkgroep geïnstalleerd om de richtlijn te herzien. De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten. De LWHB is als houder van deze richtlijn de eerst verantwoordelijke wat betreft de actualiteit van deze richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de eerstverantwoordelijke over relevante ontwikkelingen binnen hun vakgebied.