Terug naar het richtlijnenoverzicht
T cel prolymfocytenleukemie (T-PLL) is een zeldzame, agressieve T cel leukemie die bestaat uit proliferatie van kleine tot middelmatig grote T- lymfocyten, die ondanks hun post-thymus oorsprong en mature fenotype “T-prolymphocyten” worden genoemd. Slechts 2% van de chronische leukemieen bij volwassenen is T-PLL.
Gezien zeldzaamheid van ziektebeeld, wordt het wel geadviseerd bij diagnose om in ieder geval éénmalig patient/e laten beoordelen in zijn/haar referentie ziekenhuis.
Deze richtlijn is bedoeld ter optimalisering van diagnostiek, behandeling en follow-up van T-PLL en toepasbaar in alle Nederlandse ziekenhuizen die deze ziekte behandelen.
Deze richtlijn is bestemd voor alle professionals die betrokken zijn bij de diagnostiek, behandeling en begeleiding van patiënten met T-PLL, zoals internist-hematologen, en verpleegkundig specialisten
Organisatie: HOVON-CLL-werkgroep
Namen werkgroepleden richtlijn
Bellido, S. Kersting, G.D. te Raa, R. Raijmakers, H. van der Straaten, L. Tick, K. de Heer, F. Kleijwegt, M. van Gelder, J.K. Doorduijn
Belangenconflict: De HOVON-CLL-werkgroep verricht studies met financiële ondersteuning van de volgende firma’s: Abbvie, Acerta, Janssen
S. Kersting: vergoeding voor bijwonen/spreken congres: Celgene. Ontvangen research gelden: Roche, Abbvie, Janssen
M. van Gelder: Vergoedingen voor consulting van Advisory Board: Janssen, Roche, Gilead, Celgene, Abbvie
Vergoeding voor consulting: Mundipharma
Vergoeding voor bijwonen / spreken symposium: Janssen, Roche, Gilead, Celgene
Ontvangen researchgelden: Celgene, Roche
K. de Heer: geen
M. Bellido: geen
J.K. Doorduijn: vergoeding congresbezoek: Celgene en Roche
R.A.P. Raijmakers: geen
G.D. te Raa: Vergoedingen voor consulting van Advisory Board: Abbvie
F.S. Kleijwegt: geen
H.M. van der Straaten: geen
L. Tick: geen
Kwaliteitsindicatoren
Kwaliteitsindicator diagnostiek bij diagnose
Bloedonderzoek:
Kwaliteitsindicator behandeling 1
Behandelindicatie en behandelplan conform advies richtlijn
Kwaliteitsindicator behandeling 2
De conceptrichtlijn is voor commentaar aangeboden aan de leden van de Nederlandse Vereniging voor Hematologie (NVvH). Het commentaar geeft input vanuit het veld om de kwaliteit en de toepasbaarheid van de richtlijn te optimaliseren en landelijk draagvlak voor de richtlijn te genereren. Circa [5] respondenten maakten van deze mogelijkheid gebruik. Alle commentaren werden vervolgens beoordeeld en verwerkt door de werkgroep. De richtlijn werd op [15-01-2021] door de HOVON-CLL-werkgroep inhoudelijk vastgesteld. Ten slotte is de richtlijn ter autorisatie gestuurd naar de Nederlandse Vereniging voor Hematologie.
In de verschillende fasen van de ontwikkeling van het concept van de richtlijn is zoveel mogelijk rekening gehouden met de implementatie van de richtlijn en de daadwerkelijke uitvoerbaarheid van de aanbevelingen.
Om het gebruik in de dagelijkse praktijk te bevorderen wordt deze richtlijn verspreid onder de professionals van de bij de totstandkoming van deze richtlijn betrokken organisatie(s):
Module 1 Diagnostiek T-PLL
Welk diagnostisch onderzoek dient verricht te worden bij verdenking T-PLL?
Anamnese: B symptomen, klachten passend bij anemie, hepato-splenomegalie of pleuravocht, ascites, huidafwijkingen, aanwijzing voor centraal zenuwstelsel lokalisatie.
Lichamelijk onderzoek: grootte van lymfklieren, lever en milt, huidafwijkingen, aanwijzing voor pleuravocht of ascites.
Bloedonderzoek:
Immunofenotypering perifeer bloed (tabel 1)
Op indicatie (indien nodig om diagnose te stellen, indien bijvoorbeeld geen kenmerkende lokalisatie):
Moleculair onderzoek
Cytogenetisch onderzoek: Karyotypering, FISH
Op indicatie (indien verdenking andere oorzaken van pancytopenie andere dan T-PLL infiltratie):
Beenmergonderzoek: cytomorfologie, immunofenotypering, histologie en immuunhistochemie.
Diagnostische criteria T-PLL
|
Major criteria |
|
>5 x 10^9/L cellen met T-PLL fenotype in bloed of beenmerg |
|
T-cel clonaliteit aangetoond (met TCRB en TCRG genherschikkingsonderzoek of flowcytometrie) |
|
Afwijkingen van 14q32 of Xq28 OF expressie van TCL1A/B of MTCP1** |
|
Minor criteria |
|
Afwijkingen van chromosoom 11: (11q22.3; ATM) |
|
Afwijkingen in chromosoom 8: idic(8)(p11), t(8,8), trisomie 8q |
|
Afwijkingen in chromosoom 5, 12, 13, 22, of complex karyotype |
|
Kenmerkende lokalisatie van T-PLL (zoals splenomegalie, pleuravocht of ascites) |
|
Diagnose T-PLL |
|
3 major criteria |
|
Eerste 2 major criteria en 1 minor criterium |
** gevallen zonder TCL1A-, TCL1B- of MTCP1-herschikking of hun respectievelijke overexpressie worden verzameld als “TCL1-familie negatieve T-PLL”
Welk diagnostisch onderzoek dient verricht te worden vooraf aan behandeling van T-PLL?
Anamnese: Niveau van functioneren (WHO performance score) co-morbiditeit
Lichamelijk onderzoek: vastleggen grootte van lymfklieren, lever en milt, huidafwijkingen, pleuravocht of ascites.
Bloedonderzoek:
Beeldvorming: CT hals, thorax, abdomen
Module 2 Prognosticering T-PLL
Welke onderzoeken moeten gedaan worden om prognose in te schatten bij T-PLL?
Geen
Module 3 Stadiering van T-PLL
Wanneer is er een behandelindicatie voor T-PLL?
Vaststellen van actieve ziekte (tabel 3)
Module 4 Behandeling van T-PLL
Wat is de eerstelijns behandeling van T-PLL
Aanbevelingen
Alemtuzumab intraveneus zo mogelijk gevolgd door allogene stamceltransplantatie
Doserings schema:
Week 1:
Begeleidende medicatie:
Week 2-16 (of tot maximale respons):
Co-medicatie: PCP profylaxe tot minstens 3 maanden na alemtuzumab behandeling en CMV profylaxe tot 6 maanden na alemtuzumab behandeling.
Alemtuzumab kan infuusreacties veroorzaken: gradering volgens CTCAE laatste versie, v.5: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf
Bij alemtuzumab gebruik, bloedbeeld, diff, ALAT, ASAT, LDH, CMV PCR, EBV PCR wekelijks monitoren. Ook bestraalde bloedproducten toedienen.
Wanneer de behandeling langer dan 7 dagen wordt onderbroken moet de dosis alemtuzumab opnieuw worden opgebouwd
Zo mogelijk includeren in een klinische studie
Aanbevelingen
Anamnese: niveau van functioneren (‘WHO performance’-score), B symptomen, klachten passend bij anemie, hepato-splenomegalie of pleuravocht, huidafwijkingen, aanwijzing voor centraal zenuwstelsel lokalisatie
Lichamelijk onderzoek: vastleggen van grootte van lymfklieren, lever en milt, huidafwijkingen, ascites of pleuravocht
Bloedonderzoek: Hb, leukocyten, trombocyten, leukocytendifferentiatie met aantal lymfocyten, LDH
Anamnese: niveau van functioneren (‘WHO performance’-score), B symptomen, klachten passend bij anemie, hepato-splenomegalie of pleuravocht, huidafwijkingen, aanwijzing voor centraal zenuwstelsel lokalisatie, bijwerking van behandeling.
Lichamelijk onderzoek: vastleggen van grootte van lymfklieren, lever en milt en hoeveelheid pleuravocht of ascites, huidafwijkingen.
Bloedonderzoek: Hb, leukocyten, trombocyten, leukocytendifferentiatie met aantal lymfocyten, LDH
Beenmergonderzoek: (alleen indien noodzakelijk om een complete remissie te bevestigen)
Beeldvorming:
CT hals, thorax, abdomen (bij vermoeden op CR en 4-6 weken na einde behandeling)
Aanbevelingen
Anamnese: B symptomen, klachten passend bij anemie, hepato-splenomegalie of pleuravocht, ascites, huidafwijkingen, aanwijzing voor centraal zenuwstelsel lokalisatie.
Lichamelijk onderzoek: grootte van lymfklieren, lever en milt, huidafwijkingen, aanwijzing voor pleuravocht of ascites.
Bloedonderzoek:
Immunofenotypering perifeer bloed (tabel 1)
Op indicatie (indien nodig om diagnose te stellen, indien bijvoorbeeld geen kenmerkende lokalisatie):
Moleculair onderzoek
Cytogenetisch onderzoek: Karyotypering, FISH
Op indicatie (indien verdenking andere oorzaken van pancytopenie andere dan T-PLL infiltratie):
Beenmergonderzoek: cytomorfologie, immunofenotypering, histologie en immuunhistochemie.
Tabel 1: Immunofenotypering bij T-PLL bij diagnose
|
|
Percentage patienten |
|
cyTCL1+ |
>90% |
|
CD3+ |
>80% |
|
CD4+ |
60% |
|
CD5+ |
100% |
|
CD7+ |
>90% |
|
CD8+ |
15% |
|
CD4+ CD8+ |
25% |
|
CD52++ |
100% |
Tabel 2: Diagnostische criteria T-PLL
|
Major criteria |
|
>5 x 10^9/L cellen met T-PLL fenotype in bloed of beenmerg |
|
T-cel clonaliteit aangetoond (met TCRB en TCRG genherschikkingsonderzoek of flowcytometrie) |
|
Afwijkingen van 14q32 of Xq28 OF expressie van TCL1A/B of MTCP1** |
|
Minor criteria |
|
Afwijkingen van chromosoom 11: (11q22.3; ATM) |
|
Afwijkingen in chromosoom 8: idic(8)(p11), t(8,8), trisomie 8q |
|
Afwijkingen in chromosoom 5, 12, 13, 22, of complex karyotype |
|
Kenmerkende lokalisatie van T-PLL (zoals splenomegalie, pleuravocht of ascites) |
|
Diagnose T-PLL |
|
3 major criteria |
|
Eerste 2 major criteria en 1 minor criterium |
** gevallen zonder TCL1A-, TCL1B- of MTCP1-herschikking of hun respectievelijke overexpressie worden verzameld als “TCL1-familie negatieve T-PLL”
Onderbouwing
De klinische presentatie van T-PLL bestaat uit B-symptomen, hepato-splenomegalie en meestal extreme lymfocytose >100 x 10 9/L. Nodale en extra-nodale presentatie komt ook vaak voor, waaronder huid, pleurale of peritoneale effusie in ongeveer 25% van de patiënten en centraal zenuwstelsel betrokkenheid in minder dan 10% van de patiënten. Periorbitaal/conjunctivaal oedeem en perifeer oedeem komt vaak voor.
Er zijn 3 morfologische varianten van T-PLL in bloed of beenmerg. In 75% is er sprake van middelgrote cellen met een hoge kern/cytoplasma ratio, “moderatly condensed chromatin, een zichtbare nucleolus en een licht basofiel cytoplasma zonder granulae maar met typische cytoplasmatische uitstulpingen (blebs). In 20% is er een kleine cel variant met “condensed chromatin” en een nucleolus die niet zichtbaar is met lichtmicroscopie. In ongeveer 5% zijn de kernen onregelmatig en lijken op de ceribriforme kernen van Sezary of mycosis fungoides cellen.
T-PLL cellen zijn post thymus T cellen met co-expressie van mature T-lymphoide markers en vaak met cytoplasmatische expressie van TCL1 oncogenen. (tabel 1)
T-PLL dient onderscheiden te worden van andere leukemische T-cel neoplasieën zoals Sezary syndroom, ‘T-cel Large granular lymphocyte leukemia’ en ‘adult T-cell lymphocytic leukemia’, of andere rijpe T-cel tumors met leukemische presentatie.
Voor bevestiging van de diagnose is aantonen van klonale rearrangement van T-cel receptor genen (TRB of TRG) noodzakelijk. Bij T-PLL is er vaak een complex karytoype met specifieke genetische afwijkingen, die kunnen helpen bij het stellen van de diagnose.
Bij meer dan 90% van de patiënten zijn karakteristieke kenmerken van een grote populatie clonale T cellen en genetische afwijkingen van TCL1A, TCL1B (TML1) of MTCP aanwezig. (tabel 2) Bij een klein gedeelte van de patiënten zijn deze genetische afwijkingen er niet (zogenaamde TCL1-family negatieve T-PLL) en is het voor het stellen van de diagnose noodzakelijk dat een andere genetische afwijking geassocieerde aan T-PLL aanwezig is of een typische klinische lokalisatie (minor criteria).
Literatuurverantwoording
Er is gebruik gemaakt van de ondergenoemde richtlijnen zonder aanvullende systematische literatuur-analyse:
Aanbevelingen
Anamnese: Niveau van functioneren (WHO performance score) co-morbiditeit
Lichamelijk onderzoek: vastleggen grootte van lymfklieren, lever en milt, huidafwijkingen, pleuravocht of ascites.
Bloedonderzoek:
Beeldvorming: CT hals, thorax, abdomen
Onderbouwing
Om een inschatting te maken welke behandelopties mogelijk zijn bij de patiënt is het belangrijk het niveau van functioneren en comorbiditeiten in kaart te brengen. Aanvullend bloed onderzoek is er opgericht om inzicht te krijgen in prognostische markers (LDH, b2 microglobuline) en eventuele actieve of chronische infecties die kunnen verergeren door de behandeling met monoklonale antistoffen. Beenmergonderzoek en CT scan zijn nodig om adequate responsbeoordeling mogelijk te maken. De waarde van PET/CT in T-PLL is nog niet onderzocht.
Literatuurverantwoording
Er is gebruik gemaakt van de ondergenoemde richtlijnen zonder aanvullende systematische literatuur-analyse:
Aanbevelingen
Geen
Onderbouwing
Net als bij andere lymfoproliferatieve aandoeningen kunnen verhoogd LDH en beta 2 microglobuline een weergave zijn van de ziekte last en geassocieerd zijn met verminderde overleving.
In een retrospectieve studie van de MD Anderson waren verschillende factoren geïdentificeerd voorspellend voor een significant verhoogd risico op overlijden (aanwezigheid van pleurale effusie, Hb <5.7 mmol/L, hoog LDH >1668 EH/L) en voor een significant hoger risico op ziekteprogressie na initiële therapie (patiënten met niet-blanke etniciteit, afwezigheid van kleincellige variant van T-PLL en hoge b2M (8mg/l).
Mutaties in JAK3, aangetoond met next generation sequencing hebben een negatieve invloed op prognose. Aanvullend onderzoek is nodig om de waarde in de dagelijkse praktijk hiervan vast te stellen.
Literatuurverantwoording
Er is gebruik gemaakt van de onderstaande richtlijn zonder aanvullende systematische literatuur-analyse:
Wanneer is er een behandelindicatie voor T-PLL?
Aanbevelingen
Vaststellen van actieve ziekte (tabel 3) (SORT C)
Onderbouwing
Net als bij chronische lymfatische leukemie, is er pas een behandelindicatie bij actieve ziekte.
Het merendeel van de patiënten heeft bij diagnose typische klachten van hepato-splenomegalie, gegeneraliseerde lymfadenopathie en hoog aantal lymfocyten (klassiek > 100 x 10^9/L). Echter, ongeveer 20-30% van de patiënten presenteert zich aanvankelijk met stabiele of langzaam progressieve ziekte. Bij deze patiënten kan afgewacht worden met behandeling tot er actieve ziekte is, aangezien behandeling in een vroeger stadium de prognose niet verandert. (tabel 3).
Een van de onderstaande criteria is voldoende voor actieve ziekte:
Tabel 3: Criteria voor actieve ziekte
|
Constitutionele symptomen |
Significante vermoeidheid: WHO performance score ≥2 Ongewild gewichtsverlies > 10% in 6 maanden Nachtzweten zonder aanwijzing voor infectie Koorts > 38OC zonder aanwijzing voor infectie |
|
Symptomatisch beenmergfalen |
Hb < 6.2 mmol/l trombocyten < 100 x 109/l |
|
Snelle groei van lymfklieren, lever of milt |
>50% in 2 maanden, verdubbeling binnen 6 maanden Symptomatisch vergrote klieren, lever of milt |
|
Progressieve lymfocytose |
Indien > 30 x 109/l: > 50% < 2 maanden; lymfocyten verdubbelingstijd < 6 maanden |
|
Extranodale betrokkenheid |
Orgaan infiltratie, pleuravocht of ascites, Centraalzenuwstelsel lokalisatie |
Literatuurverantwoording
Er is gebruik gemaakt van de onderstaande richtlijn zonder aanvullende systematische literatuur-analyse:
Aanbevelingen
Alemtuzumab intraveneus zo mogelijk gevolgd door allogene stamceltransplantatie2
Doserings schema:
Week 1:
Begeleidende medicatie:
Week 2-16 (of tot maximale respons):
Co-medicatie: PCP profylaxe tot minstens 3 maanden na alemtuzumab behandeling en CMV profylaxe tot 6 maanden na behandeling.
Alemtuzumab kan infuusreacties veroorzaken: gradering volgens CTCAE laatste versie, v.5:
Bij alemtuzumab gebruik, bloedbeeld, diff, ALAT, ASAT, LDH, CMV PCR, EBV PCR wekelijks monitoren. Ook bestraalde bloedproducten toedienen.
Wanneer de behandeling langer dan 7 dagen wordt onderbroken moet de dosis alemtuzumab opnieuw worden opgebouwd
Onderbouwing
De beste eerstelijns behandeling om complete remissie te behalen is intraveneus alemtuzumab (anti-CD52) met een respons kans van >90%. Er is voorkeur voor alemtuzumab intraveneus in plaats van alemtuzumab subcutaan gezien de resultaten van de pilot UKCLL05 studie: ORR 91% vs 33% respectievelijk met een mortaliteit tijdens eerstelijnsbehandeling van 22% met het gebruik van alemtuzumab subcutaan. Vanwege de slechte resultaten van alemtuzumab subcutaan werd de studie eerder stopgezet. Na inductie met alemtuzumab intraveneus is de mediane remissieduur echter kort, deze is minder dan 2 jaar. Voor patiënten in complete remissie dient allogene stamceltransplantatie dan ook overwogen te worden. Ongeveer een derde van de patiënten bereikt hiermee langdurige overleving. Een conditionering met totale lichaamsbestraling met een dosis van tenminste 6 Gy is geassocieerd met een lagere kans op recidief. Indien er geen allogene donor beschikbaar is kan autologe stamceltransplantatie een optie zijn, echter zonder kans op langdurige overleving.
Literatuurverantwoording
Er is gebruik gemaakt van de onderstaande richtlijn zonder aanvullende systematische literatuur-analyse:
Aanbevelingen
Zo mogelijk includeren in een klinische studie
Onderbouwing
Er is geen consensus over een tweedelijns therapie. Herhaalde behandeling met alemtuzumab, indien de T-PLL bij recidief CD52 + blijft (CD52 kan negatief worden na eerdere behandeling met alemtuzumab) leidt in de helft van de patiënten tot opnieuw remissie, maar deze is meestal van korte duur. De effectiviteit en veiligheid van venetoclax en ibrutinib zal in Nederland onderzocht worden voor deze indicatie.
Literatuurverantwoording
Er is gebruik gemaakt van de onderstaande richtlijn zonder aanvullende systematische literatuur-analyse:
Aanbevelingen
Anamnese: niveau van functioneren (‘WHO performance’-score), B symptomen, klachten passend bij anemie, hepato-splenomegalie of pleuravocht, huidafwijkingen, aanwijzing voor centraal zenuwstelsel lokalisatie
Lichamelijk onderzoek: vastleggen van grootte van lymfklieren, lever en milt, huidafwijkingen, ascites of pleuravocht
Bloedonderzoek: Hb, leukocyten, trombocyten, leukocytendifferentiatie met aantal lymfocyten, LDH
Onderbouwing
Bij patiënten met een niet actieve T-PLL dient maandelijks door middel van anamnese, lichamelijk onderzoek en bloedonderzoek vastgesteld te worden of er actieve ziekte is ontstaan. Binnen 1-2 jaar ontstaat bij bijna alle patiënten een behandelindicatie.
Literatuurverantwoording
Er is gebruik gemaakt van de onderstaande richtlijn zonder aanvullende systematische literatuur-analyse:
Aanbevelingen
Anamnese: niveau van functioneren (‘WHO performance’-score), B symptomen, klachten passend bij anemie, hepato-splenomegalie of pleuravocht, huidafwijkingen, aanwijzing voor centraal zenuwstelsel lokalisatie, bijwerking van behandeling.
Lichamelijk onderzoek: vastleggen van grootte van lymfklieren, lever en milt en hoeveelheid pleuravocht of ascites, huidafwijkingen.
Bloedonderzoek: Hb, leukocyten, trombocyten, leukocytendifferentiatie met aantal lymfocyten, LDH
Beenmergonderzoek: (alleen indien noodzakelijk om een complete remissie te bevestigen)
Beeldvorming:
CT hals, thorax, abdomen (bij vermoeden op CR en 4-6 weken na einde behandeling)
Onderbouwing
Respons beoordeling is afhankelijk van of een patiënt continue of intermitterende behandeling krijgt. Beoordeling dient elke 4-6 weken plaats te vinden. Met nieuwe continue behandelingen kan stabilisatie van een eerdere snel progressieve ziekte als een gunstige respons beoordeeld worden. In eerste instantie kan de beoordeling op basis van anamnese, lichamelijk onderzoek en bloedonderzoek gedaan worden. Bij vermoeden van complete remissie kan dit bevestigd worden met beenmergonderzoek en CT scan. De CT-scan dient dan met RECIL criteria beoordeelt te worden, waarbij eendimensionale metingen van 3 doel laesies bepaald worden. Beoordeling is net als bij CLL verdeelt in 2 soorten parameters: groep A parameters (B symptomen en mate van tumor last) en groep B parameters (effect van T-PLL op beenmergfunctie). (tabel 5)
De klinische waarde van minimale residuele restziekte detectie met immunofenotypering of moleculaire bepalingen bij complete remissie is nog niet bekend.
Tabel 4: Respons beoordeling bij behandeling T-PLL
|
Groep |
Parameter |
CR (alle aanwezig) |
CRi (3 van A aanwezig en ≥1 van B afwezig) |
PR (≥2 van A en ≥1 van B) |
SD (alle aanwezig) minimaal 3 maanden aanhouden |
PD (≥1 van A of B aanwezig) |
|
A |
lymfklieren |
Lange as diameter < 1 cm op CT |
Lange as diameter < 1 cm op CT |
Vermindering ≥30% van SLD van max. 3 doel laesies |
Verandering van -<30% tot + ≥20% |
1) Toename > 20% in SLD van max. 3 doel laesies 2) Of bij LK <1.5 cm, toename >5 mm en SLD >1.5 cm 3) Of nieuwe laesie |
|
milt |
< 13 cm |
< 13 cm |
Vermindering van ≤50% in verticale lengte van normaal ten opzichte van start |
Verandering van -49% tot +49% ten opzichte van normaal van start |
Toename ≥50% in verticale lengte vanaf normaal ten opzichte van start |
|
|
B symptomen |
geen |
geen |
-* |
– |
– |
|
|
Lymfocyten aantal |
< 4 x 109/L |
< 4 x 109/L |
≤30 x 109/L en vermindering van ≥ 50% van start |
>30 x 109/L of verandering van-49% tot +49% |
Toename ≥50% ten opzichte van start |
|
|
T-PLL cellen in Beenmerg |
T-PLL cellen <5% van mononucleaire cellen |
T-PLL cellen <5% van mononucleaire cellen |
– |
– |
– |
|
|
Extra nodale lokalisatie |
geen |
geen |
– |
– |
– |
|
|
B |
Trombocyten |
≥ 100 x 109/L zonder transfusie of TPO |
Aanwezigheid of trombopenie en/of anemie en/of neutropenie door toxiciteit van behandeling |
≥ 100 x 109/L of toename ≥50% ten opzichte van start zonder transfusie of TPO |
verandering van-49% tot +49% |
Daling ≥50% ten opzichte van start door T-PLL BM infiltratie |
|
Hemoglobine |
≥ 6,83** mmol/L zonder transfusie of EPO |
Aanwezigheid of trombopenie en/of anemie en/of neutropenie door toxiciteit van behandeling |
≥ 6,83 mmol/L of toename ≥50% ten opzichte van start zonder transfusie of EPO |
<6,83 mmol/L of <50% ten opzichte van start of toename van < 1.24 mmol/L |
Daling van ≥ 1.24 mmol/L ten opzichte van start door T-PLL BM infiltratie |
|
|
Neutrofielen |
≥ 1,5 x 109/L zonder G-CSF |
Aanwezigheid of trombopenie en/of anemie en/of neutropenie door toxiciteit van behandeling |
≥ 1,5 x 109/L of toename ≥50% ten opzichte van start zonder G-CSF |
verandering van-49% tot +49% |
Daling ≥50% ten opzichte van start door T-PLL BM infiltratie |
*- telt niet voor de definitie van PR, SD of PD
** conversiefactor Hb g/dl naar mmol/L x 0.6206
SLD: som van lange as diameter
LK: lymfklier
BM: Beenmerg
TPO: trombopoëitine
EPO: erytropoëitine
G-CSF: koloniestimulerende factoren
Literatuurverantwoording
Er is gebruik gemaakt van de onderstaande richtlijn zonder aanvullende systematische literatuur-analyse:
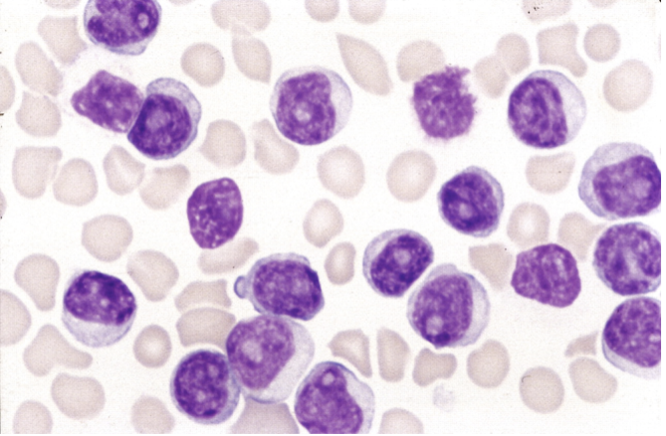
A. In 75% is er sprake van middelgrote cellen met een hoge kern/cytoplasma ratio, “moderatly condensed chromatin, een zichtbare nucleolus en een licht basofiel cytoplasma zonder granulae maar met typische cytoplasmatische uitstulpingen (blebs).
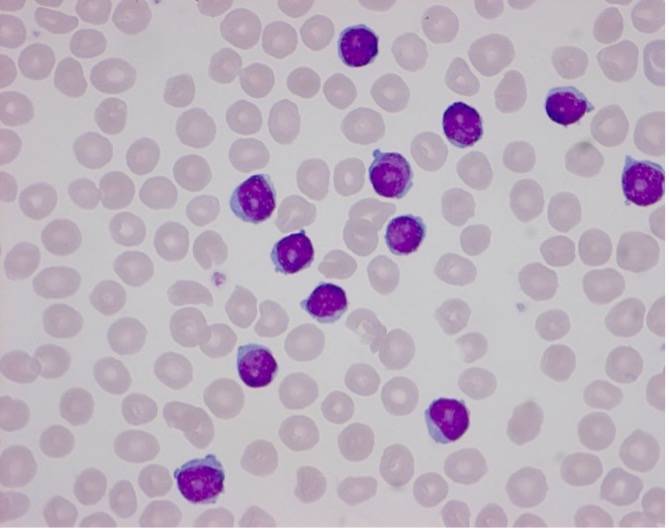
B. In 20% is er een kleine cel variant met “condensed chromatin” en een nucleolus die niet zichtbaar is met lichtmicroscopie.
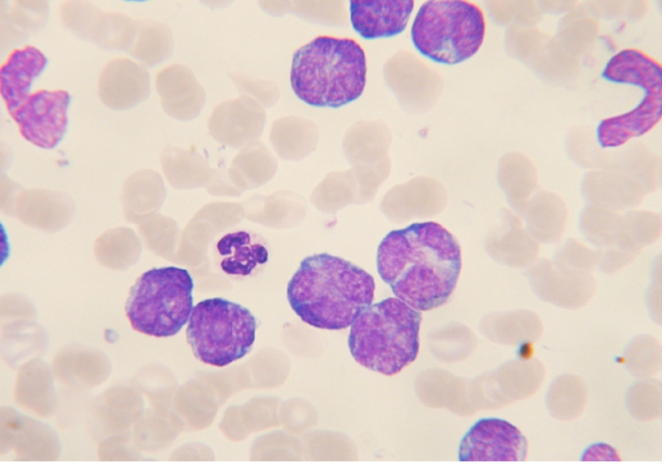
C. In ongeveer 5% zijn de kernen onregelmatig en lijken op de ceribriforme kernen van Sezary of mycosis fungoides cellen
© 2022. Alle rechten voorbehouden
Nederlandse Vereniging voor Hematologie (NVvH)