Aanbeveling
|
Conclusies |
SORT Grade |
|
De meest gangbare technieken om β-thalassemie vast te stellen zijn de HPLC en capillaire elektroforese (EC) methode. Met name ten behoeve van antenatale diagnostiek is aanvullende mutatie analyse geïndiceerd. |
A |
|
Voor de diagnostiek van α-thalassemie is altijd moleculaire diagnostiek aangewezen. In het algemeen kan volstaan worden met het bepalen van de 7 meest voorkomende deleties met GAP-PCR. |
A |
|
De belangrijkste algemene lab parameters bij de diagnostiek van β-thalassemie zijn: Een microcytaire (MCV > 50 en < 70 tot max 80 fL), hypochrome (verlaagd MCH) anemie, met een verhoogd aantal reticulocyten en in de perifere rode cel uitstrijk schietschijf cellen, diverse andere vormafwijkingen en erytroblasten, zonder dat sprake is van een verlaagde ijzerstatus. |
A |
|
De belangrijkste algemene lab parameters bij de diagnostiek van α-thalassemie zijn: Een microcytaire (MCV > 50 en < 70 tot max 80 fL), hypochrome (verlaagd MCH) anemie, met een verhoogd aantal reticulocyten. |
A |
Onderbouwing
Introductie
Er zijn diverse fysiologische vormen van hemoglobine. De aanwezigheid van deze vormen kan verschillen per leeftijd en per individu.
In de postnatale situatie zijn deze vormen: HbA (bestaande uit 2 α- en 2 β – ketens), HbA2 (2 α- en 2 δ-ketens) en HbF (2 α- en 2 γ- ketens).
In de fysiologische situatie bevatten de rode bloedcellen van een volwassene ongeveer 97-98% HbA, 2-3% HbA2 en een minimale hoeveelheid HbF en van een pasgeborene ongeveer 70 -95% HbF, 5-30% HbA en < 1% HbA2.
Gedurende het eerste levensjaar neemt de expressie van het HbF af en wordt in de fysiologische situatie geleidelijk aan vervangen door HbA expressie.
Thalassemie
Bij thalassemie is sprake van verminderde of afwezige globine keten productie.
Afhankelijk van de verminderde of afwezige productie van het soort keten, is bijvoorbeeld sprake van α-, β-, γ-, δ-thalassemie of combinaties hiervan.
De belangrijkste vormen van thalassemie zijn α- en β-thalassemie. Deze erven autosomaal recessief over en de ernst van het ziektebeeld kan uitgedrukt worden als klinisch beeld (fenotype) of als genotype (soort mutatie).
Mensen met een heterozygote genetische variant van α- of β-thalassemie zijn veelal asymptomatisch en behoeven geen behandeling.
Mensen met een homozygote of samengesteld heterozygote genetische thalassemie variant, vertonen in wisselende mate klinische expressie, waarvan bloedarmoede de belangrijkste is.
Tegenwoordig spreken we van TDT (transfusie afhankelijke thalassemie) en NTDT (niet- transfusie afhankelijke thalassemie) (zie Figuur 1).
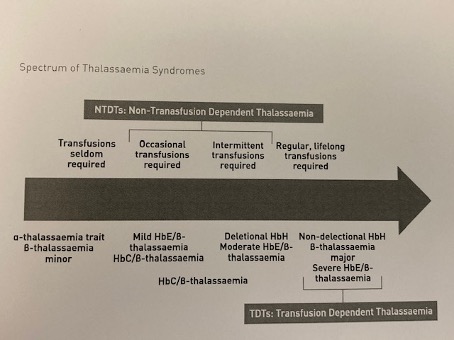
Figuur 1: Spectrum van de thalassemie syndromen.
Diagnostiek van β-thalassemie
Normaal gesproken heeft een individu één actief β-gen op elke kopie van chromosoom 11. Er zijn tot op heden meer dan 200 mutaties in het β-gen beschreven, aanleiding gevend tot variabele klinische expressie. Een β0-mutatie houdt in dat er geen enkele expressie door het betreffende gen plaatsvindt en dus geen β-globine productie. Een β+-mutatie houdt in dat er verminderde expressie door het betreffende gen plaatsvindt en dus verminderde β-globine productie (rond 10%). Bij β++-mutaties is de verminderde productie erg mild.
De belangrijkste algemene laboratorium bevindingen bij β-thalassemie zijn (zie ook Figuur 2): Een microcytaire (MCV > 50 en < 70 tot max 80 fL), hypochrome (verlaagd MCH) anemie, met een verhoogd aantal reticulocyten en in de perifere rode cel uitstrijk schietschijf cellen, diverse andere vormafwijkingen en erytroblasten, zonder dat sprake is van een verlaagde ijzerstatus.
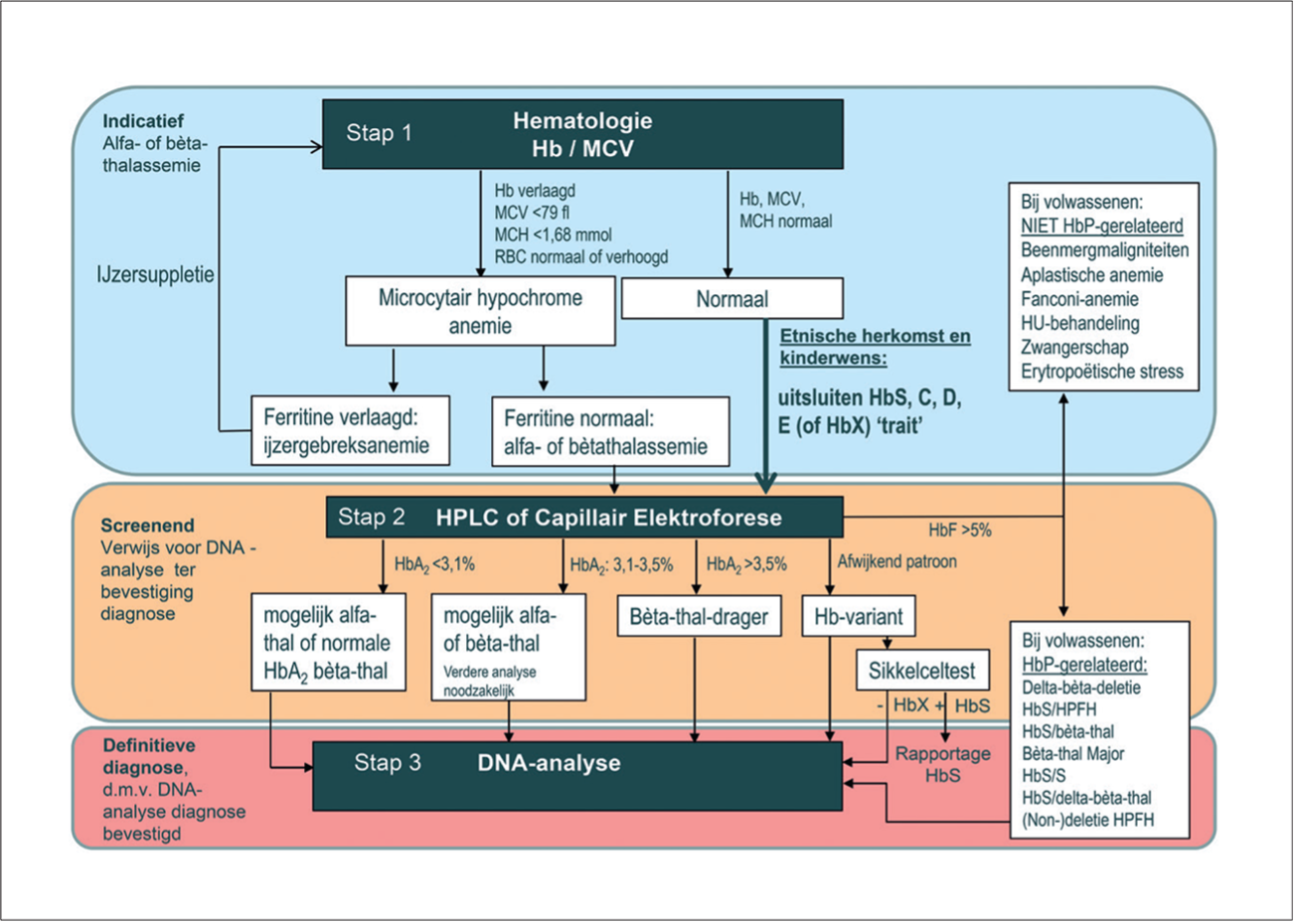
Figuur 2. Stroomdiagram diagnostisch proces voor hemoglobinopathie en dragerschap hiervan.
Hemoglobine analyse met kwantitatieve technieken dient ingezet te worden ter diagnostiek en bestaat uit:
Met deze kwantitatieve technieken wordt de hoeveelheid en type aanwezig hemoglobine bepaald.
Bij kinderen vanaf 1 jaar en volwassenen met homozygote β0-thalassemie is er geen aanwezigheid van HbA, een verhoogd gehalte HbA2 en sprake van een HbF gehalte tussen 92 en 95%.
Voor pasgeborenen met homozygote β0-thalassemie geldt: geen HbA, HbF > 95%, HbA2 < 2.5%.
Bij kinderen vanaf 1 jaar en volwassenen met homozygote β+-thalassemie of samengestelde β+/β0-mutaties, is het HbA gehalte tussen 10 en 30%, de HbA2 waarde verhoogd en de HbF waarde tussen 70 en 90%.
Voor pasgeborenen met deze intermedia vormen geldt: HbA < 5%, HbF > 95% en HbA2 < 2.5%.
Moleculaire analyse
Aanvullende moleculaire analyse is in het algemeen niet noodzakelijk voor het stellen van de juiste diagnose, maar kan in sommige gevallen zeker van diagnostische waarde zijn. In geval van prenatale diagnostiek is dit noodzakelijk om vast te kunnen stellen of het ongeboren kind al dan niet aangedaan is.
Veel voorkomende mutaties kunnen met PCR technieken vastgesteld worden. Als met deze gerichte mutatie analyse geen mutatie aangetoond kan worden, kan het aangewezen zijn om β-globine gen sequentie analyse uit te voeren.
Genotype-fenotype correlatie
De disbalans in globine ketens (α- versus β-ketens) vormt een belangrijke determinant van de ernst van het klinisch beeld van de β-thalassemie. Om deze reden zijn factoren die deze disbalans verminderen, mede bepalend voor het fenotype. Zo draagt een bijkomende α-thalassemie bij aan een vermindering van de ernst van de anemie. Ook mutaties (o.a. de HPFH mutatie) die aanleiding geven tot verhoogde productie van γ-ketens, hebben een dergelijk gunstig effect.
Aan de andere kant kan het fenotype in negatieve zin worden beïnvloed door de aanwezigheid van een toegenomen keten disbalans door toename van α-ketens als gevolg van een toegenomen aantal functionele α- globine genen (triplicatie of quadruplicatie).
Diagnostische workup β-thalassemie in samenvatting
Diagnostiek van α-thalassemie
Normaal gesproken heeft elk individu 4 α-genen ( 2 gedupliceerde α-genen op 2 loci) op de korte arm van chromosoom 16.
In tegenstelling tot β-thalassemie, wordt bij α-thalassemie de verminderde of afwezige synthese van de ketens veelal veroorzaakt door deleties, optredend in het α-globine gen cluster op chromosoom 16. Er zijn ruim 125 verschillende moleculaire defecten beschreven die α-thalassemie kunnen veroorzaken, die sterk in grootte verschillen. De meest voorkomende deleties zijn de South East Asian (-SEA) deletie, die 2 α-genen betreft en de 4.2 en 3.7 kb-deleties.
De verschillende α-thalassemie fenotypes zijn het gevolg van het ontbreken van één (α+-thalassemie) of beide (α0-thalassemie) α-globine genen op elk van de beide loci.
De belangrijkste algemene laboratorium bevindingen bij de klinisch belangrijke vormen van α-thalassemie (HbH ziekte) zijn (zie ook Figuur 2):
Moleculaire analyse
Voor het stellen van de diagnose α-thalassemie is moleculaire diagnostiek aangewezen. In veel laboratoria beperkt deze diagnostiek zich tot de 7 meest voorkomende deleties, die met behulp van GAP-PCR vastgesteld kunnen worden. Bij persisterende verdenking op α-thalassemie na uitsluiten van deze deleties, kan uitgebreidere moleculaire diagnostiek met α-gen sequencing of MLPA (multiplex ligation-dependent probe amplification) verricht worden in daartoe gespecialiseerde laboratoria. Ter opsporing van de non-deletionele vormen, die ook oorzaak kunnen zijn van instabiliteit van de α-ketens, wordt vaak de techniek van geautomatiseerde Direct-Sequencing technieken volgens Sanger gebruikt.
HbH ziekte is de klinisch belangrijkste vorm van α-thalassemie en kan het gevolg zijn van het ontbreken van 3 α-genen of van een combinatie van deletionele en non-deletionele mutaties, waarvan Hb Constant Spring de meest voorkomende is (zie Tabel 1). De non-deletionele vorm van HbH ziekte is geassocieerd met een ernstiger fenotype.
Ook kunnen extra α-genen een β-thalassemie intermedia fenotype veroorzaken bij dragers van β-thalassemie. Alfa-triplicatie (de anti alfa 3.7) kan via een PCR gebaseerde methode worden aangetoond of d.m.v. MLPA.
Tabel 1. Genotype-fenotype correlatie bij α-thalassemie
|
Fenotype |
Genotype |
Kliniek/bloedbeeld |
|
Silent carrier |
α0α/αα |
Asymptomatisch Geen hematologische afwijkingen |
|
Drager/minor |
α0α/α0α α0α0/αα |
Milde asymptomatische anemie Microcytose en hypochromie |
|
Deletionele HbH ziekte |
α0α0/α0α |
Milde tot matige anemie Niet-transfusie afhankelijk Kliniek varieert van minor tot major |
|
Non-deletionele HbH ziekte |
α0α0/αTα |
Matige tot ernstige anemie Kan incidentele of frequente transfusies nodig hebben |
|
Major (HbBart’s hydrops foetalis) |
α0α0/α0α0 |
De meesten ontwikkelen hydrops foetalis en overlijden intrauterien of kort post partum Indien overleving (antenatale transfusies), dan transfusie afhankelijk |
De ‘T’ staat voor een non-deletionele α keten mutatie, zoals Hb Constant Spring
Bij pasgeborenen kan HbH ziekte vermoed worden door het aantonen met Hb-typering (o.a. met HPLC) van HbBart’s. HbBart’s bestaat uit 4 γ-ketens en ontstaat in afwezigheid van voldoende α-ketens. Dit HbBart’s verdwijnt echter snel na de geboorte. HbBart’s van rond 25% bij een pasgeborene suggereert de aanwezigheid van HbH ziekte.
Op latere leeftijd kan bij Hb-typering HbH (4 β-ketens) aangetoond worden, echter dit is niet bij alle patiënten met α-thalassemie aanwezig.
HbBart’s hydrops foetalis wordt veroorzaakt door de deletie van alle 4 α-genen en is in principe niet met het leven verenigbaar (tenzij het kind antenataal en reeds vroeg in de zwangerschap bloedtransfusies ontvangt). Indien een kind met HbBart’s overleeft, dan zal het ook postnataal transfusie afhankelijk blijven.
Genotype-fenotype correlatie
De mate van anemie is verschillend voor de diverse vormen van HbH ziekte: bij de deletionele vorm variërend tussen 4.3 en 6.6 mmol/L; bij de non-deletionele vorm tussen 2.4 en 5.4 mmol/L.
Diagnostische workup α-thalassemie in samenvatting
Zoekverantwoording
Er is onder andere gebruikt gemaakt van bestaande richtlijnen.
Referenties
Aanbevelingen
De volgende endocriene onderzoeken zijn geïndiceerd:
|
Monitoring endocriene parameters |
Eerste keer op leeftijd van |
Frequentie |
|
Gewicht, lengte, zithoogte |
vanaf diagnose |
1x/jaar tot eindlengte bereikt |
|
Evaluatie groeicurve |
vanaf diagnose |
1x/jaar |
|
TSH, vrij T4* |
Vanaf 5 jaar (TDT), 10 jaar (NTDT) |
1x/jaar |
|
Tanner stadia |
vanaf 10 jaar |
2x/jaar |
|
Anamnese menarche, menstruatiecyclus |
Vanaf 10 jaar |
1x/jaar |
|
Testosteron |
M, na puberteit |
1x/jaar |
|
Nuchter glucose |
Vanaf 10 jaar |
1x/jaar |
|
Orale glucose tolerantie test |
Vanaf puberteit |
1x/jaar |
|
Ochtend cortisol (om 8:00) |
Vanaf puberteit |
1x/jaar |
|
Vitamine D |
Vanaf 2 jaar |
1x/jaar |
|
Serum calcium, fosfaat |
Vanaf 10 jaar |
1x/jaar |
|
PTH |
Bij volwassenen |
1x/jaar |
|
Zink Koper, Selenium, Cerulopasmine, Vitamine A, C, E |
Bij TDT, chelatie |
1x/jaar |
|
Botdensitometrie (BMD lumbale wervelkolom en heup) |
Vanaf eind puberteit/voor transitie |
A 2 jaar
|
|
Op indicatie |
|
|
|
IGF1-1, IgF-BP3, X-hand, Verder conform consensus kleine lengte |
Indien groeivertraging |
|
|
Groeihormoonprovocatietest |
Bij groeivertraging (na behandelen van evt. hypothyreoïdie, deficiëntie voeding) |
|
|
Consult diëtiste |
Indien groeivertraging |
|
|
PTH |
Wanneer serum calcium verlaagd |
|
|
FSH, LH, oestradiol (V), testosteron (M), X-hand |
Indien vertraagde puberteit (nog niet gestart op leeftijd 13 jaar (V), 14 jaar (M)) |
|
* Een (secundaire) bijnierschors insufficiëntie dient uitgesloten te worden voor start suppletie van schildklierhormoon.
2.
3.
|
Bisfosfonaten |
Route toediening |
Dosis
|
|
Alenodroninezuur |
per os |
10 mg 1 dd of 70 mg 1x/week |
|
Pamidroninezuur |
parenteraal |
30 mg /maand |
|
Zoledroninezuur |
Parenteraal |
4 mg/ 3 maanden (TIF guideline) 5 mg/jaar (Farmacotherapeutisch Kompas) |
|
Neridroninezuur |
Parenteraal |
100 mg / 6 maanden |
Gebruik bisfosfonaten altijd in combinatie met Vit D en Calcium suppletie. Na een behandelingsduur van 12 maanden dient een BMD-meting verricht te worden, waarna de vervolgbehandeling wordt afgestemd. Zoledroninezuur en neridroninezuur zijn middelen van eerste keuze voor thalassemie geassocieerde osteoporose (Guisti A et al., 2016, Piga et al. 2017).
Intraveneuze toediening van zoledroninezuur schijnt effectiever te zijn dan orale bisfosfonaten.
De behandeling van osteoporose bij kinderen gebeurt altijd in overleg met een kinderendocrinoloog.
Onderbouwing
Inleiding
Verhoogde ijzerabsorptie door ineffectieve erytropoëse en chronische hemolytische anemie veroorzaken ijzerstapeling o.a. in de pancreas, hypofyse en bijschildklierklieren en kunnen leiden tot endocriene dysfunctie zoals diabetes mellitus, hypogonadisme, hypoparathyreoïdie en infertiliteit (zie hiervoor hoofdstuk zwangerschap en fertiliteit).
De behandeling met bloedtransfusies bij TDT leidt tot een onderdrukking van de ineffectieve erytropoëse, waardoor er verschil is in prevalentie van endocrinopathieën in TDT en NTDT. De prevalentie in NTDT is relatief lager, echter stijgt deze met toegenomen leeftijd.
Splenectomie, de mate van ineffectieve erytropoëse en een laag foetaal Hb gehalte worden geassocieerd met endocriene dysfuncties in NTDT. Het gebruik van Hydrea lijkt hierin een positieve rol te spelen. Bij diagnose van een hypofysaire hormoon deficiëntie is het belangrijk extra alert te zijn op mogelijke uitval van andere hypofyse-assen.
|
Conclusie |
SORT Grade |
|
1.Management van endocriene complicaties is complex. Bij afwijkende monitoring dient de (kinder-)endocrinoloog laagdrempelig te worden betrokken. |
C |
|
2.A. Start zo vroeg mogelijk met chronische transfusies en ijzerchelatie om beenmerghyperplasie en ijzertoxiciteit te voorkomen. Zie hiervoor de aanbevelingen hoofdstuk transfusie, ijzerstapeling en ontijzering. |
A |
|
2.B. Houd je aan het maximale doseeradvies van Deferoxamine bij kinderen (40 mg/kg/dag) |
B |
|
2.C. Let op een adequate inname van calcium (700-1000 mg per dag) |
B |
|
2.D. Streef Vitamine D spiegels van ~ 50 nmol/l na |
B |
|
2.E. Geef leefstijladviezen zoals voldoende lichamelijke activiteit, zonlicht exposure, vermijden van roken en vraag voor dieetadviezen laagdrempelig een diëtist in consult |
B |
|
2.F. Probeer diabetes mellitus vroegtijdig op te sporen |
B |
|
3.A. Overweeg bij volwassenen een behandeling met bisfosfonaten wanneer er sprake is van: a. een BMD Z-score < -2.0 (pre-menopauze, < 50 jr.), T-score < -2.5 (postmenopauze, > 50 jr.) of b. fragility fracturen en/of afname van de botdichtheid ondanks adequate Vitamine D-spiegel of c. bij het gebruik van hormoonbehandeling in het kader van hypogonadisme. |
B |
|
3.B. Gebruik bisfosfonaten altijd in combinatie met Vit D en Calcium suppletie. Na een behandelingsduur van 12 maanden dient een BMD meting verricht te worden, waarna de vervolgbehandeling wordt afgestemd. |
B |
|
3.C. Zoledroninezuur en neridroninezuur zijn middelen van eerste keuze voor thalassemie geassocieerde osteoporose (Guisti A et al., 2016, Piga 2017). Intraveneuze toediening van zoledroninezuur schijnt effectiever te zijn dan orale bisfosfonaten. Zie bvg tabel voor keuze en dosering bisfosfonaten. |
B |
|
3.D. Bij kinderen duiden vertebrale compressiefracturen ontstaan zonder hoogenergetisch trauma of lokale ziekte op osteoporose. In afwezigheid van vertebrale compressiefracturen wordt osteoporose bij kinderen gedefinieerd als:
N.B. Bij de interpretatie van de DXA-scan dient rekening gehouden te worden met eventuele kleine lengte en/of achterlopende skeletrijping (bv bij late puberteit). Een methode om de botdichtheid te corrigeren voor de kleine lengte is het berekenen van de Bone Mineral Apparent Density (Ward et al; Arch Dis Child. 2007 Jan;92(1):53-9. doi: 10.1136/adc.2006.097642). |
A,B |
|
3.E. De behandeling van osteoporose bij kinderen gebeurt altijd in overleg met een kinderendocrinoloog. |
C |
Samenvatting literatuur
Verhoogde ijzerabsorptie door ineffectieve erythropoiese en chronische hemolytische anemie veroorzaken ijzerstapeling o.a. in de pancreas, hypofyse en bijschildklierklieren, en kunnen leiden tot endocriene dysfunctie zoals diabetes mellitus, hypogonadisme, hypoparathyreoïdie en infertiliteit.
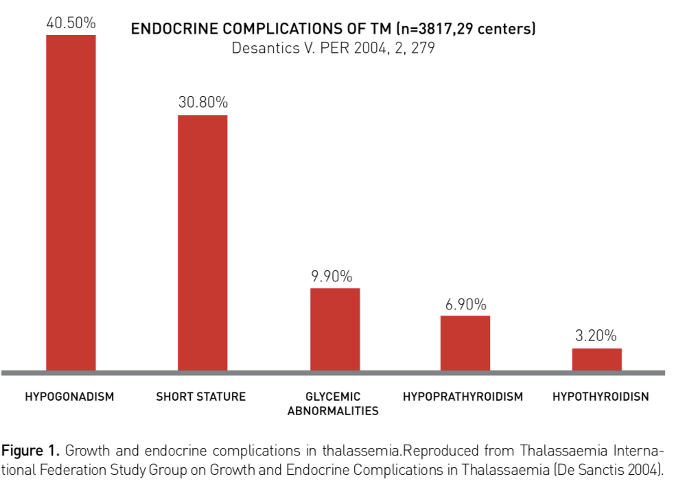
Late puberteit en hypogonadisme
Late puberteitsontwikkeling en hypogonadotroop hypogonadisme zijn de meest voorkomende klinische gevolgen van ijzerstapeling in de hypofyse. Gonadale ijzerstapeling komt veel minder vaak voor. Bij meisjes spreekt men van vertraagde puberteit bij ontbreken van borstontwikkeling op de leeftijd van 13 jaar, bij jongens als de testes op de leeftijd van 14 jaar nog < 4 ml zijn. Een stagnatie in de puberteitsontwikkeling is een relatief vaak voorkomende complicatie in patienten met TDT met matige tot ernstige ijzerstapeling. Hierbij is er gedurende een jaar geen progressie van puberteitsstadia en een afname van de groeisnelheid. Bij meisjes kan hypogonadotroop hypogonadisme zich verder uiten als primaire of secundaire amenorroe. Prevalentie van hypogonadisme in adolescenten en volwassenen met thalassemie major is 38% bij vrouwen en 43% bij mannen (guideline TDT, Cappelini et al.). Geadviseerde routineonderzoeken zijn een X-hand en BMD-meting, bepaling van LH, FSH en testosteron. Patiënten met thalassemie hebben doorgaans hypogonadotroop hypogonadisme gekenmerkt door lage gonadotrofinen en oestradiol/testosteron.
Behandeling (door kinderarts-endocrinoloog): Doel is het bewerkstelligen van ontwikkeling van puberteitskenmerken, optimaliseren van de lengtegroei en voorkomen van complicaties van hypogonadisme zoals osteoporose.
Bij late puberteit kan in eerste instantie gekozen worden voor een kortdurende behandeling van ongeveer 6 maanden en hierna de behandeling te evalueren via bloedcontrole van de hypothalamus-hypofyse-gonadale as.
Meisjes: Oestrogeenbehandeling per os of transdermaal. Afhankelijk van de leeftijd, lengtegroei en het spontaan bereikte puberteitsstadium zal de startdosis worden bepaald en een schema voor ophogen tot een volwassen onderhoudsdosis. Twee jaar na start puberteitsinductie wordt een progestageen toegevoegd.
Jongens: Testosteronbehandeling intramusculair of transdermaal. Afhankelijk van de leeftijd, lengtegroei en het spontaan bereikte puberteitsstadium zal de startdosis worden bepaald en een schema voor ophogen tot een volwassen onderhoudsdosis.
Hypothyroidie
Hypothyroidie als complicatie bij TDT is voornamelijk geassocieerd met ijzerstapeling en is ongebruikelijk in patienten met een optimaal chelatieregime. De frequentie van hypothyroïdie varieert van 4 – 29% en lijkt gerelateerd aan genotype, leeftijd, ethnische variatie en verschil in behandeling (transfusie frequentie en ontijzeringstherapie) (de Sanctis et al., 2019). Zowel primaire hypothyreoidie als centrale hypothyreoidie komen voor. Monitoring van schildklierfunctie (vrij T4 en TSH) wordt jaarlijks geadviseerd vanaf een leeftijd van 5 jaar bij TDT en 10 jaar bij NTDT. Zonodig zullen thyroid auto-antistoffen worden bepaald in combinatie met een echografie van de schildklier.
Gradatie van schildklier-dysfuncties:
Subklinische hypothyroidie is een combinatie van een hoog TSH met een normaal vrij T4. De combinatie van een hoog TSH met een laag vrij T4 kenmerkt klinische primaire hypothyroidie. De klassieke klinische presentatie van hypothyroidie in thalassemia major is lastig te herkennen omdat symptomen niet specifiek zijn en gerelateerd kunnen worden aan de anemie. Ernstige hypothyroïdie in TM kan zich uiten met een ernstige groeiachterstand met vertraagde puberteit, vertraagde botrijping, cardiaal falen en pericardeffusie (de Sanctis 2013). Centrale hypothyreoïdie wordt gekenmerkt door een verlaagd FT4 met een laag of inadequaat normaal (soms mild verhoogd) TSH.
De behandeling is afhankelijk van de ernst van de hypothyroidie. Een goede compliance van de ontijzeringstherapie kan hypothyroidie voorkomen danwel verbeteren in geval van milde subklinische hypothyroïdie (TSH 5 -10 mU/L). In het geval van manifeste hypothyreoïdie is de behandeling met L-thyroxine. Aandacht behoeft de patient met subklinische vorm van hypothyroidie en cardiomyopathie: behandeling met amiodaron (antiaritmica klasse III) kan leiden tot snelle verergering van de hypothyroidie, wat weer leidt tot achteruitgang van de cardiale functie (de Sanctis et al., 2019, Alexandrides 2000).
Afwijkende OGTT en diabetes mellitus (DM)
Een afwijkende OGTT en diabetes mellitus zijn een veel voorkomende complicatie in TM patienten zonder optimale ontijzeringsbehandeling. Echter kan diabetes ook voorkomen bij patienten met een regelmatig transfusie regime en goede adherence van chelatietherapie. Er spelen dus ook andere bijkomende factoren een rol zoals de individuele sensitiviteit tot ijzerstapeling, chronische anemie, zink deficientie en een verhoogde collageendepositie secundair aan de verhoogde activiteit van het enzym protocollageen proline hydroxylase, dat ijzer afhankelijk is en leidt tot een verstoorde microcirculatie in de pancreas.
DM bij TM is zeldzaam gedurende de eerste tien levensjaren. Vanaf de leeftijd van 10 jaar kan een afwijkende OGTT voorkomen. De combinatie van puberteit en thalassemie geassocieerde risicofactoren geven een gedeeltelijke verklaring voor de toename van insuline resistentie in adolescenten met thalassemie.
De etiologie van DM is multifactorieel (genetische factoren, insuline deficiëntie, insulineresistentie en leverdysfunctie secundair aan virale hepatitis). Ijzerstapeling door chronische transfusies geeft schade aan de pancreatische b-cellen met verminderde insulinesecretie als gevolg, zie fig. 3., TIF-guideline.
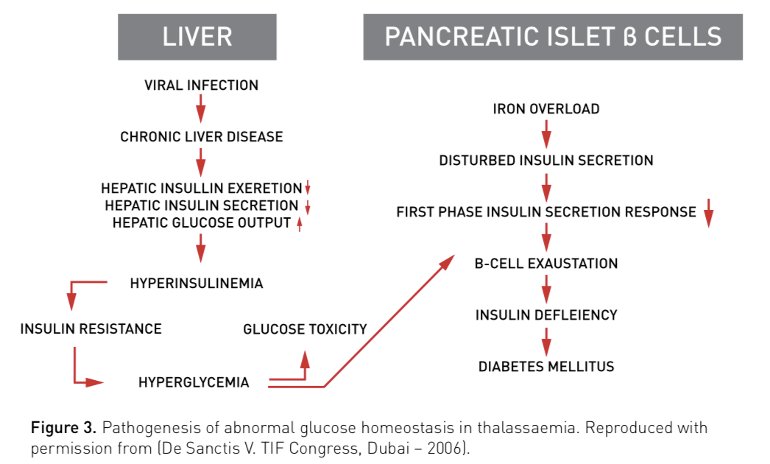
Wanneer nuchter serum glucose > 6.1 mmol/L bij jaarlijkse screening is ook een OGTT geïndiceerd. Screening door middel van een 2 h OGTT wordt geadviseerd vanaf puberteit (a 2 jaar).
Diagnostische criteria voor OGTT:
Ijzerdepositie in het pancreas is de sterkste predictor voor b-cel-toxiciteit. Pancreas T2* meting is, naast het bepalen van nuchter glucose, een mogelijke toegevoegde screeningsmethode. Verhoogde ijzerdepositie in het pancreas is een voorspeller voor een afwijkende OGTT en kan identificatie van mogelijke hoog risicopatiënten bevorderen voor dat irreversibele pancreatische schade ontstaat (Noetzli 2009, Pepe et al., 2020). Echter blijft de OGTT de gouden standaard voor evaluatie van de glucose homeostase. Screening op mogelijke hepatitis en een goede adherence chelatietherapie zijn belangrijke preventieve maatregelen.
In patienten met thalassemie en diabetes is de prevalentie van diabetische nefropathie 13-55% (Tzoulis et al. 2014, Loebstein et al. 1998) en 13-26% heeft diabetische retinopathie (Tzoulis et al., 2014, Incorvaia et al. 1998). Macrovasculaire complicaties zijn zeldzaam echter is het risico op cardiale complicaties, hartfalen, hyperkinetische arryhtmie en myocardiale fibrose bij patienten met thalassemie en diabetes significant verhoogd (Pepe et al. 2013).
Management van een afwijkende OGTT en DM zijn:
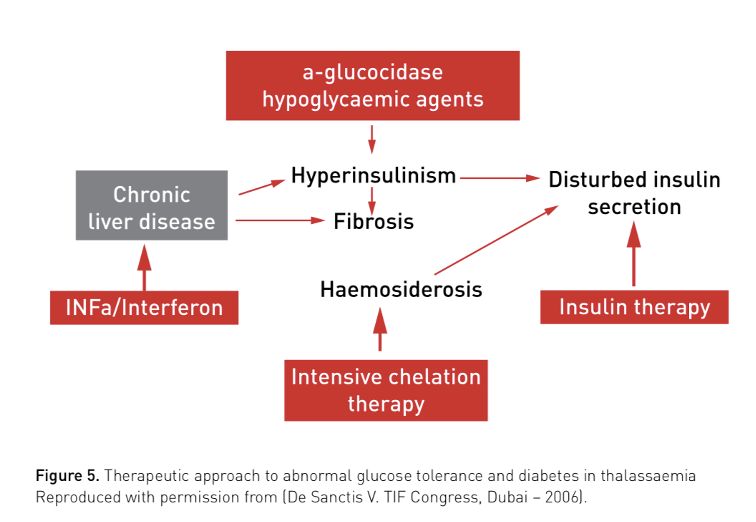
Monitoring van DM in thalassemie patiënten is niet anders dan bij de reguliere diabetes patiënt: Het HbA1c is echter niet betrouwbaar vanwege de verkorte levensduur van de erythrocyt, de ineffectieve erythropoiesis en de afhankelijkheid van chronische transfusies (de Sanctis 2013).
Hypoparathyreoïdie (HPT)
HPT is een typische complicatie in de 2e levensdecade van TDT patiënten en wordt toegeschreven aan ijzerstapeling in de bijschildklieren. De incidentie varieert van 1.2% tot 19% en verschilt per behandelcentrum. HPT lijkt vaker voor te komen bij mannen (man/vrouw ratio = 1.35) (TIF-guideline, TDT). De meerderheid van de patiënten laat een milde vorm van HPT zien met milde hypocalciemie en daardoor paresthesie. Ernstige symptomen zijn tetanie, convulsies of cardiaal falen (Skordis 2013).
Vanaf de leeftijd van 10 jaar (TDT-guideline adviseert 16 jaar?) wordt jaarlijks bepaling van serum calcium en serum fosfaat aanbevolen. Beoordeling in combinatie met Vitamine D-status en zonodig PTH.
Behandeling gebeurt in overleg met een kinderendocrinoloog en bestaat uit orale toediening van calcium en actief vitamine D om serum calcium spiegels te normaliseren. Soms zijn hiervoor hoge doses nodig, cave hypercalciemie bij overbehandeling (met risico op ontwikkeling van nefrolithiasis, nefrocalcinose). Calcitriol 0.25-1.0 ug 2 dd is meestal voldoende om calcium en fosfaat spiegels te normaliseren. Een calcium rijk en fosfaat arm dieet kan worden geadviseerd.
Bijnierinsufficiëntie
Prevalentie van biochemische centrale bijnierinsufficientie onder thalassemie patienten varieert van 0-45% (El Kholy 2013). Symptomen kunnen soms moeilijk worden herkend doordat spierzwakte, artralgie, gewichtsverlies regelmatig bij thalassemie patiënten voorkomen.
Het bepalen van ochtendcortisolspiegel (nuchter tussen 8-9 uur) wordt een a twee jaarlijks geadviseerd in patienten met een groeihormoondeficientie. Zonodig moet een ACTH test worden verricht.
Osteoporose (ook wel TBD ‘thalassemia bone disease’)
Osteoporose is een veel voorkomende oorzaak van morbiditeit in patienten met TDT danwel NTDT en komt voor in 40-50% van adequaat behandelde TDT-patiënten.
Gezien de ernstige toxiciteit van ijzerstapeling heeft een adequate ijzerchelatie hoogste prioriteit in de behandeling en preventie van osteoporose.
De pathogenese van osteoporose in thalassemie is complex en multifactorieel. Naast de ontwikkeling van botdistorsie door ineffectieve erythropoëse en progressieve beenmergexpansie zijn er een aantal genetische en verworven factoren, welke leiden tot een dysbalans in bot remodeling door inhibitie van osteoblasten activiteit en een toegenomen osteoclasten activiteit met als gevolg botdestructie. Veel patienten met thalassemie laten een verlaagde botmineraaldichtheid (BMD) zien, welke kan worden gemeten door middel van een DEXA-scan. Een evt verlate puberteit en hypogonadisme spelen hierin een oorzakelijke rol. Ook wordt een verhoogd fractuurrisico beschreven, waarbij de relatie tussen een lage BMD en het risico op fracturen niet geheel duidelijk is. Een verlaagde botdichtheid kan ook bij kinderen met thalassemie (< 10 jaar) worden gezien. Er is echter geen evidence voor behandeling van een lage BMD bij kinderen in de afwezigheid van klinisch significante fracturen, derhalve weinig onderbouwing om een BMD-meting te verrichten voor beëindiging van de puberteit. Bij kinderen bestaat management voornamelijk uit life style interventies zoals adequate calcium intake (suppletie van 700 – 1000mg/dag vanaf leeftijd 11 jaar), adequate Vit D-spiegels en voldoende lichamelijke activiteit.
Zonder Vitamine D-suppletie is vitamine D deficiëntie in thalassemie patiënten zeer gebruikelijk. Vitamine D is essentieel voor calcium homeostase en mineralisatie van het skelet. Vit D-spiegels laten een directe correlatie zien met de botdichtheid. Er wordt gestreefd naar Vit D-spiegels van – 50 nmol/l.
Risicofactoren voor thalassemia bone disease zijn oudere leeftijd, groeiretardatie, rugpijn, tekenen van zenuwcompressie, ernstige osteoporose, botbreuken. Een afname in lengte kan een aanwijzing zijn voor vertebrale fracturen.
Bij kinderen kan groeivertraging optreden door langdurig hoge doses deferoxamine (> 40 mg/kg/dag). Gezien het toenemend gebruik van deferasirox wordt dit nog maar zelden gezien.
Zoekverantwoording
Er is onder andere gebruikt gemaakt van bestaande richtlijnen.
Referenties
Aanbevelingen
Algoritme voor de diagnostiek naar pulmonale hypertensie
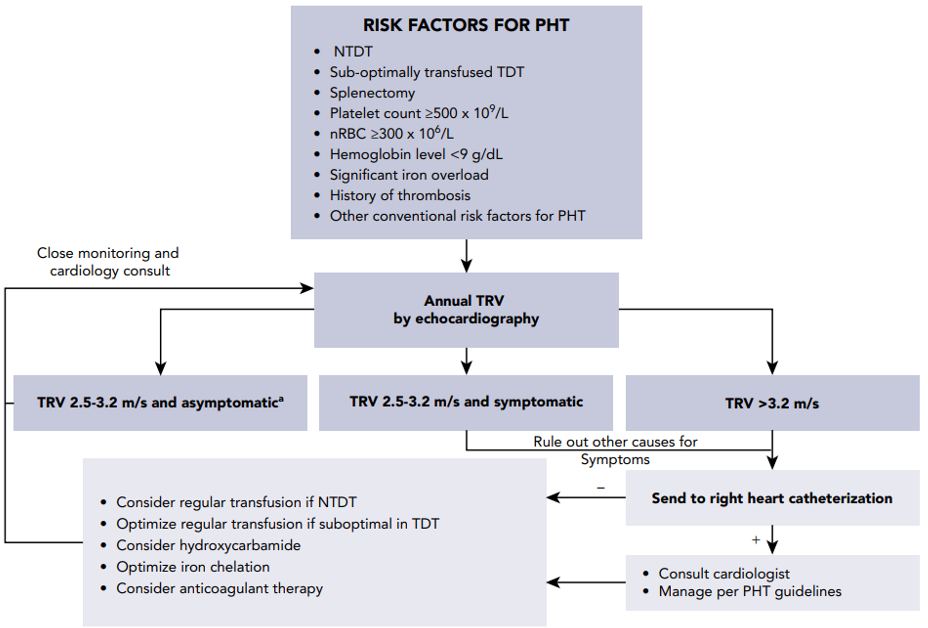
Figure 1. Beleid t.a.v. diagnose, preventie, and behandeling van PH in volwassenen met β-thalassemia. NB: Patienten met TRV < 2.5 m/s wordt echo cor na 3-5 jaar geadviseerd. nRBC, nucleated red blood cell count; PHT, pulmonary hypertension.
|
Conclusie |
score |
|
1x/3 jaar echocardiografie met TRV bepaling (vanaf 15 jaar oud, o.i eerder) |
B3 |
|
TRV 2,5 – 3,2 m/s, alleen katheterisatie bij klachten en anders echo jaarlijks herhalen |
B3 |
|
TRV ≥ 3,2 m/sec, optimalisatie transfusiebeleid en consulteer een longarts of cardioloog met PH expertise voor rechter hartkatheterisatie en overweeg behandeling |
C3 |
Onderbouwing
PH wordt gedefinieerd als een toename van de gemiddelde PAP druk > 25 mmHg gemeten met rechter hartkatheterisatie. PH bij thalassemie komt zeker voor met name bij NTDT (Vlados 2012 en Morris 2010). Studies die middels echocardiografie PH beoordelen, tonen bij 40-50% patiënten met NTDT en 10-75% bij TDT afwijkingen mogelijk passend bij PH aan (Fraidenburg ANN NYA SC 2016). . In deze studies werd ook aangetoond dat 33% van de patiënten met thalassemie een TRV 2,5-3,2 m/s heeft en 5% een TRV > 3m/s. Rechter hartcatheterisatie bevestigde bij 94% van de patiënten met een TRV > 3,2 m/s een PH, terwijl dit percentage onbekend is bij TRV tussen 2,5 en 3,2% (Derchi Circulation 2014).
Bekende voorspellende factoren voor PH bij NTDT en TDT zijn leeftijd, status na splenectomie, ernstige hemolyse, hepatitis C, verhoogde tromboseneiging of eerdere trombose en de mate van ijzerstapeling (Fraidenburg, Ann 2016). In de enige publicatie waarin rechter hartcatheterisatie de aanwezige PH bevestigde, werd geen associatie gevonden tussen de ferritine waarde en PH (derchi). In totaal betrof het hier 27 β-thalassemie patiënten.
Ook kan PH het gevolg zijn van arteriële hypertensie, veneuze hypertensie of PH van chronische longziekte of chronische trombo-embolische ziekte.
Er bestaan geen prospectieve placebo gecontroleerde trials voor de behandeling van PH bij thalassemie. Een positief effect werd gesuggereerd in NTDT van hydroxycarbamide, evt. in combinatie met L-carnitine (Fraidenburg, Amoozgar EJH 2011, Karimi EJH 2009 en EJH 2010), maar gerandomiseerde studies ontbreken. Beperkte case reports en case series zijn gemeld met sildenafil (Littera, Derchi, Correale, Morris) en prostacycline agonisten en endotheline antagonist (Anthi, Tam, Ussavarungsi en Burger).
Het valt te overwegen het transfusiebeleid aan te scherpen voor patiënten met een TRV > 2,5 m/s en symptomen passend bij PH en derhalve te streven naar volledige onderdrukking van de ineffectieve erytropoëse (Taher Blood 2010).
De waarde van NT-proBNP bij de diagnostiek van pulmonale hypertensie is niet bekend. Er is wel aangetoond dat een verhoog NT-proBNP een relatie heeft met diastolische dysfunctie (2010 D.T. Kremastinos, Cong Heartfailure, D.T Kremastinos, 2010, Am Heart J).
Zoekverantwoording
Er is onder andere gebruikt gemaakt van bestaande richtlijnen.
Referenties