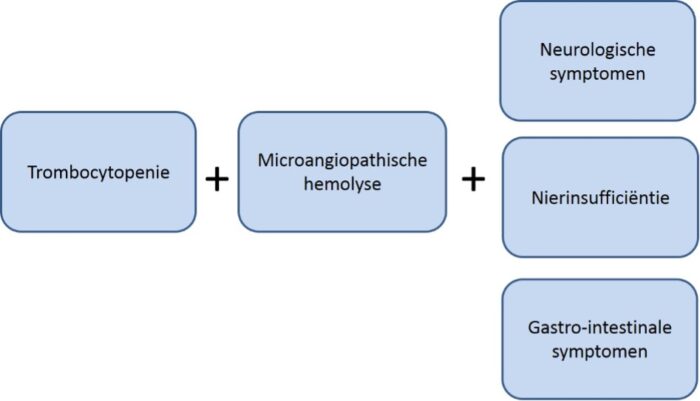
Alle vormen van trombotische microangiopathie hebben overlappende symptomatologie die optreedt door obstructie van de microcirculatie met aggregaten van trombocyten en fibrinedraden. Hierdoor treedt mechanische intravasculaire hemolyse op en worden trombocyten verbruikt, waardoor uiteindelijk orgaanschade optreedt in de organen waarin de microcirculatoire afwijkingen optreden (Figuur 1).2
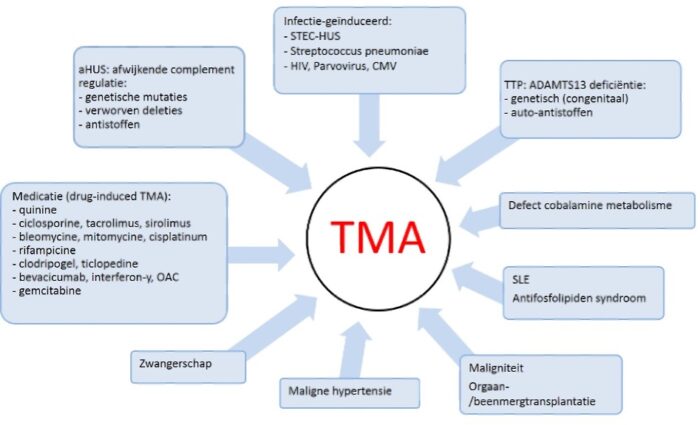
Er zijn diverse oorzaken van TMA bekend. Het klinisch beeld overlapt gedeeltelijk, maar soms is op grond van de voorgeschiedenis of bepaalde ziekteverschijnselen wel een oorzaak voor de TMA waarschijnlijk (Figuur 2). De meest bekende oorzaken zijn:
Verwarrend in de naamgeving is het feit dat de termen HUS en TTP beide het proces van trombotische microangiopathie beschrijven. Aan deze termen kan men dan ook niet de onderliggende oorzaak herkennen. Het is een kwestie van afspraak welke oorzaak-met-ziektebeeld met welk acroniem wordt aangeduid. Wij pleiten ervoor om de term TMA te gebruiken zo lang de oorzaak van de TMA niet is vastgesteld.
Belangrijke differentiaal diagnostische overwegingen bij verdenking TMA zijn: diffuse intravasale stolling, hemofagocytair syndroom, glucose-6-fosfaat-dehydrogenase-deficientie (G6PD deficiëntie), paroxysmale nachtelijke hemoglobinurie, andere vormen van hemolytische anemie.
Bij de eerste presentatie is de kliniek vaak niet altijd even duidelijk:3, 23, 24
Vaak wordt er een uitlokkende factor beschreven zoals virale infectie, vaccinatie en zwangerschap.
Ook CaHUS kent extrarenale symptomen in de acute fase (ongeveer 20%):25
CaHUS kent familiaire (15-20%) alsmede sporadische vormen en kan recidiveren. Eerste manifestaties kunnen op alle leeftijden optreden, ook bij genetische oorzaken (bijvoorbeeld in zwangerschap).
De klinische waarschijnlijkheidsdiagnose CaHUS wordt gesteld indien er geen andere oorzaken voor TMA aanwezig zijn, dat wil zeggen geen STEC infectie, ADAMTS13 activiteit ≥10% en geen andere onderliggende oorzaken van secundaire TMA.1, 3
Verlaagd complement C3 kan een aanwijzing zijn voor CaHUS, maar wordt lang niet bij alle patiënten beschreven. Een normaal serum C3 sluit de diagnose CaHUS derhalve niet uit. Het meten van de complement eiwitten C3/C4 wordt wel ingezet, maar heeft geen discriminerende functie in de differentiaal diagnose. Een verlaagd C3 kan wel een aanwijzing zijn voor CaHUS. Een verlaagd C4 is niet typisch voor CaHUS.
CaHUS ontstaat ten gevolge van een ongecontroleerde activatie van de alternatieve route van het complement systeem (zie figuur 4).
Dit alles maakt dat men tegenwoordig spreekt over een genetisch complement profiel, zogenaamd complementtype met een verhoogde vatbaarheid voor CaHUS.
Genetische analyse van complementgenen alsmede aantonen van de aanwezigheid van antistoffen tegen factor H wordt gedaan in het Radboudumc te Nijmegen:
en door Sanquin te Amsterdam:
Bij alle patiënten met verdenking op CaHUS dient genetische diagnostiek en analyse naar antistoffen tegen factor H ingezet te worden. Het vinden van een complement mutatie ondersteunt de diagnose, kan helpen het recidief risico in te schatten en is ook nodig bij de donorselectie rondom niertransplantatie. In overleg met de landelijke werkgroep CaHUS kan binnen 2 weken spoed-genetisch onderzoek (Radboudumc) verricht worden. Bij onduidelijke oorzaak van TMA wordt laagdrempelig overleg met lid van landelijke CaHUS werkgroep, aanwezig in ieder UMC, geadviseerd. De indicatie-commissie van de landelijke CaHUS werkgroep besluit of er gestart kan worden met eculizumab.
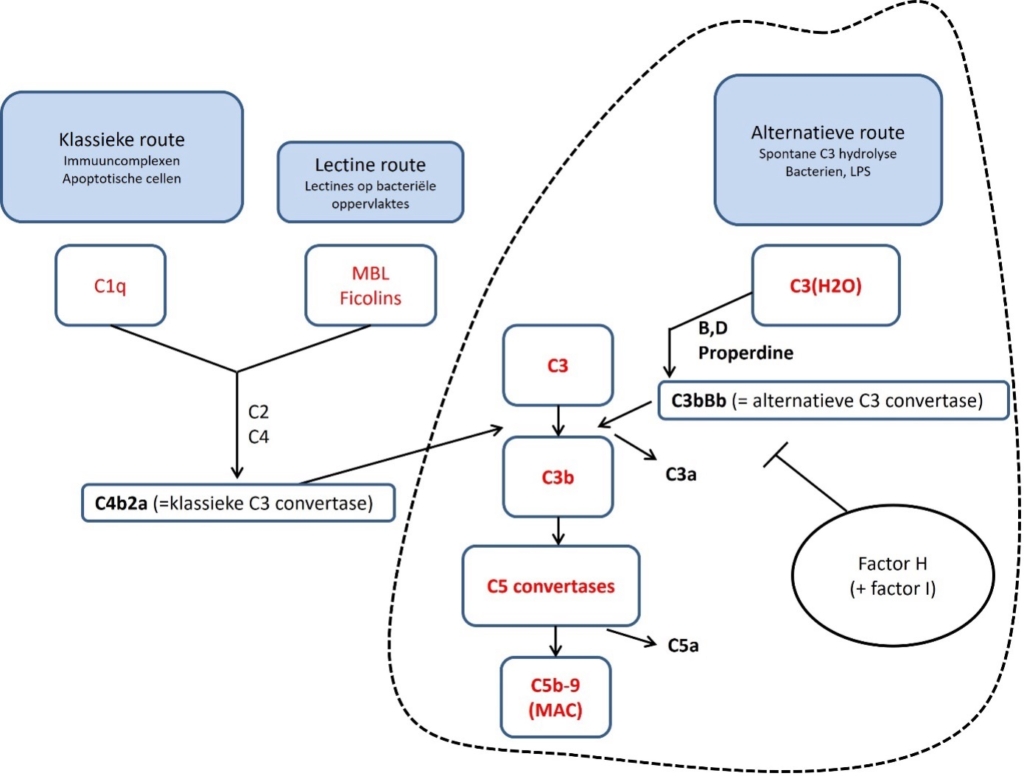
Figuur 3. Het pathofysiologisch mechanisme bij CaHUS: dysregulatie van de alternatieve route van het complement systeem door o.a. ‘loss of function’ mutaties in factor H en I of juist ‘gain of function’ mutaties in factor B en C3. Dit leidt uiteindelijk tot een persisterende activatie van de alternatieve route.
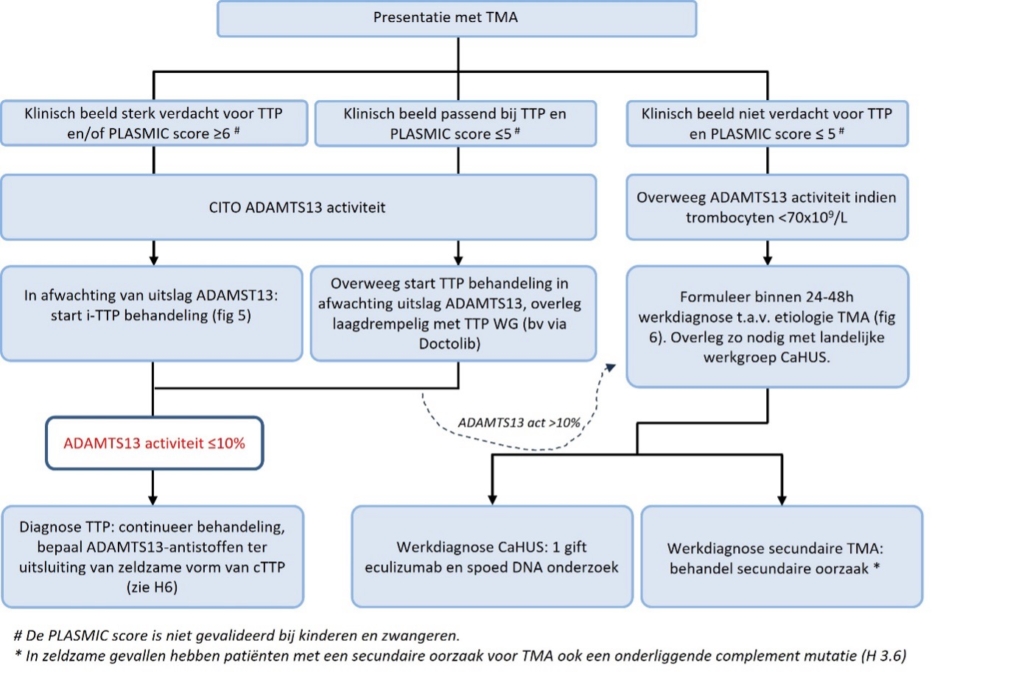
Figuur 4. Initiële therapeutische strategie bij TMA.
Bij een hoge verdenking op acute verworven (=immuun gemedieerde) TTP (i-TTP), corresponderend met een PLASMIC score van 6 of 7,8 is het noodzakelijk om direct behandeling te starten om (verdere) orgaanschade te voorkomen. Gezien het snel progressieve karakter van de aandoening dient bevestiging van een ADAMTS13 activiteit <10% niet afgewacht te worden.
De initiële behandeling van verworven TTP bestaat uit:
Figuur 5 illustreert schematisch het behandelschema van verworven TTP.
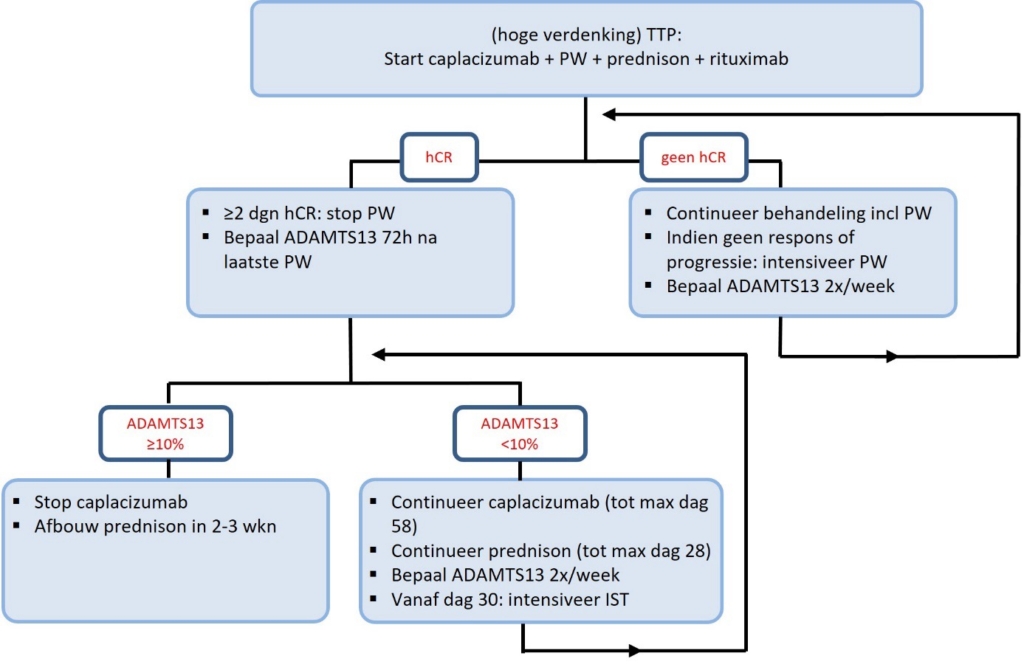
Figuur 5. Behandelschema verworven TTP.
PW = plasmawisseling. hCR = hematologische complete respons.
Plasmawisseling
Caplacizumab
Caplacizumab is een gehumaniseerd nanobody dat bindt aan het A1-domein van het von Willebrand factor (VWF) eiwit. Door deze binding wordt de interactie van VWF met het glycoproteine Ib-IX-V op de trombocyt voorkomen waardoor het micro-angiopathische proces een halt wordt toegeroepen. Op deze manier dient het middel ter overbrugging tot intreden van immunologisch effect van prednison en eventueel andere immunosuppressieve middelen. Belangrijkste bijwerking inherent aan dit mechanisme is het optreden van bloedingen, welke over het algemeen als niet-ernstig classificeren. Het risico op ernstige bloedingen bedraagt ongeveer 2% en het risico op een ernstige intracerebrale bloeding onder caplacizumab <1%.63 De richtlijncommissie is van mening dat het middel (relatief) gecontra-indiceerd is bij een TTP gepaard gaande met levensbedreigende bloedingen (WHO graad 3-4). Er ontbreekt bewijs over gebruik van caplacizumab bij patiënten met een verhoogd bloedingsrisico, zoals bij gebruik van profylactische of therapeutische antistolling of in geval van TTP-gerelateerde cerebrale ischemische haarden met (risico op) hemorrhagische transformatie. De werkgroep is van mening dat terughoudendheid hier geboden is. Met betrekking tot antistolling kan een praktische benadering zijn om caplacizumab zo snel mogelijk na coupering van antistolling te starten. Bij cerebrale bloedingen dient men zeer terughoudend te zijn; de uitgebreidheid van de cerebrale ischemie versus het bloedingsvolume en de verwachte progressie van beide onder behandeling van caplacizumab dienen hierbij tegen elkaar afgewogen te worden.64 Overleg met de landelijke TTP werkgroep wordt in deze uitzonderlijke situaties sterk geadviseerd.
Behandeling met caplacizumab tot maximaal 58 dagen na laatste plasmaferese is veilig en effectief gebleken in de eerste lijnsbehandeling van TTP.65-70 De voor plasmaferese benodigde hoeveelheid plasma neemt met meer dan 30% af, responsen treden sneller op met reductie van exacerbaties en de ziekenhuis- en opnameduur wordt verkort. Wel is de kans op een recidief TTP in de periode na staken van het middel groter indien geen immunologische complete respons (=ADAMTS13 activiteit ≥10%) is bereikt. De werkgroep is van mening dat calpacizumab als initiële behandeling aan de standaardbehandeling van plasmawisseling en corticosteroïden bij alle patiënten dient te worden toegevoegd.
Caplacizumab bij kinderen <18 jaar
Prednison
Rituximab
Overige maatregelen:
Behandeling van (verdenking op) CaHUS met eculizumab dient plaats te vinden volgens (consensus) advies van de indicatie-commissie van de landelijke werkgroep CaHUS (zie figuur 7). Overleg bij de verdenking op CaHUS met het lid van de landelijke werkgroep in het dichtstbijzijnde academische centrum. De patiënt-casus zal door het desbetreffende lid met 3 andere leden van de werkgroep (indicatie-commissie) besproken worden, waarna advies wordt gegeven over behandeling met eculizumab.
Het algoritme is gebaseerd op resultaten van prospectieve studies en expert-opinie van de werkgroep leden. De volgende uitgangspunten zijn gehanteerd:
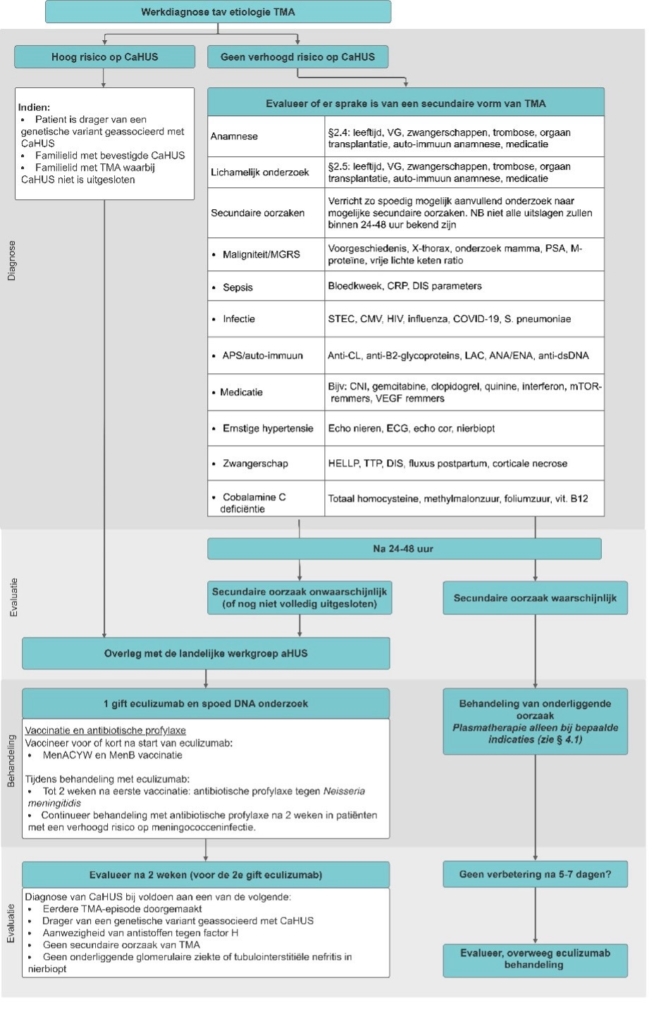
Figuur 6. Behandeling van CaHUS.
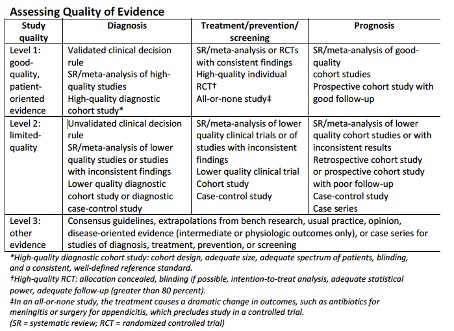
Sterkte van aanbevelingen op basis van SORT systematiek

Nadler formule totaal bloedvolume (L)48
Waarbij: Ht = hematocriet; G = gewicht (kg)
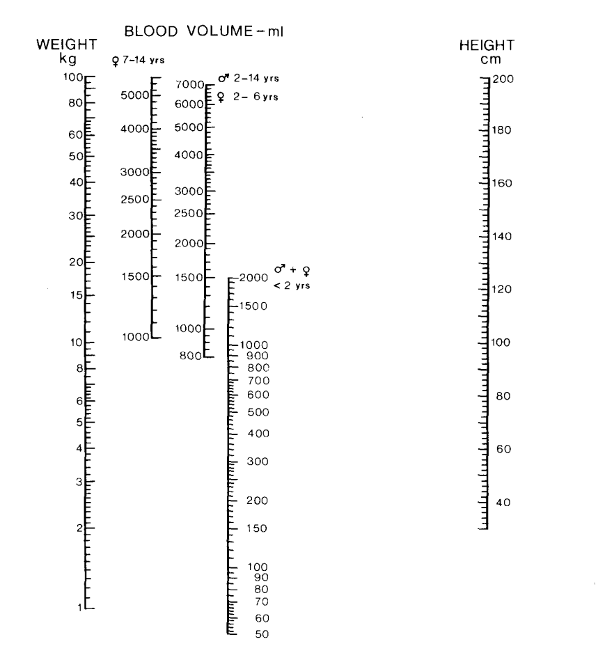
Linderkamp monogram49
Trek een lijn tussen gewicht en lengte en lees het bijbehorende bloedvolume af op de voor geslacht en leeftijd specifieke verticale balk.