Hoe is de behandeling bij een bloeding bij de ziekte van Von Willebrand?
Aanbevelingen
Onderbouwing
Inleiding
De ziekte van von Willebrand is de meest voorkomende erfelijke hemostasestoornis en wordt gekenmerkt door een tekort aan werkzaam Von Willebrand Factor (VWF). VWF heeft een dubbele rol in het bloedstollingsproces. Enerzijds bevordert het de adhesie van bloedplaatjes, anderzijds dient het als dragereiwit voor stollingsfactor VIII. Bloedingen in de huid en slijmvliezen, ten gevolge van stoornissen in de bloedplaatjesadhesie, zijn het belangrijkste klinisch kenmerk bij patiënten met VWD. Meest voorkomende bloedingen zijn menorragie (85%), hematomen (77%), nabloeden uit kleine wondjes (77%) en mondbloedingen (62%). Bij de ernstige vorm van de ziekte van von Willebrand is de factor VIII concentratie zo sterk verlaagd, dat ook bloedingen in met name spieren (20%) en gewrichten (25%) kunnen optreden. Centraal zenuwstelsel bloedingen komen niet frequent voor (2%), gastro-intestinale bloedingen daarentegen weer vaker (15%) (de Wee, 2012). Het aantal bloedingen en de ernst ervan is dus afhankelijk van de ernst van de ziekte en kan sterk variëren per individu.
Behandeling van bloedingen gebeurt, afhankelijk van de restactiviteit van VWF, door stollingsfactor correctie. Er kan gebruik gemaakt worden van VWF(/factor VIII) concentraten (plasma-derived of recombinant), of van een synthetisch preparaat, DDAVP (1-deamino-8-D-arginine-vasopressine: Minrin, intraveneus; Octostim, neusspray/subcutaan), dat tijdelijk de plasmaconcentratie van VWF/factor VIII verhoogt door een release van VWF uit de endotheelcellen te bewerkstelligen. Daarnaast wordt gebruik gemaakt van antifibrinolytica (tranexaminezuur). Voor welke behandeling en preparaat gekozen wordt, is afhankelijk van de ernst van de bloeding, het type VWD en patiënt afhankelijke factoren (zoals respons op DDAVP en eventuele contra-indicaties). Ook is het van belang dat er, indien mogelijk, lokale hemostase wordt nagestreefd.
Onder kleine bloedingen worden bijvoorbeeld verstaan: beginnende hemartros, neus- en tandvleesbloedingen, milde hematurie en bloeding na gering trauma. Ernstige bloedingen zijn bijvoorbeeld: voortgeschreden of ernstige gewrichtsbloedingen, spierbloedingen in boven- en onderarmen, kuit en musculus iliopsoas en ernstige traumata zonder manifeste bloeding. Onder levensbedreigende bloedingen worden verstaan bijvoorbeeld: schedeltrauma, tractus digestivus bloedingen, buiktrauma en bloedingen met bedreiging van de luchtweg (NVHB, 2020).
Conclusies
|
SORT Grade |
Conclusie |
|
B |
Bloedingen in de huid en slijmvliezen zijn het belangrijkste klinische kenmerk bij patiënten met VWD. (de Wee, 2012) |
|
B |
Angiodysplasieën komen met name voor bij patiënten met VWD type 2 (2%) en VWD type 3 (4.5%). (Fressinaud, 1993) |
|
C |
Gebruik van stollingsfactorconcentraat met een ratio VWF:Act/factor VIII > 10, bij patiënten met een laag factor VIII gehalte, geeft pas een stijging van het endogene factor VIII na 6-8 uur. (Borel-Derlon, 2007; Mannucci, 2013) |
Samenvatting literatuur
Resultaten
Algemeen
Factor VIII is de belangrijkste speler in chirurgische en weefselbloedingen, plaatjes afhankelijk VWF activiteit lijkt de belangrijkste speler in mucosale bloedingen (Mannucci, 2019). Behandeling van bloedingen bij patiënten met VWD bestaat dan ook uit correctie van VWF en factor VIII. Er is weinig bekend over welke waarde van VWF kritisch is voor het stoppen van een mucosale of chirurgische bloeding. De data die de aanbevelingen voor deze uitgangsvraag ondersteunen komen voort uit klinische ervaring, consensus van expert opinion en observationele studies. Voor welke behandeling en preparaat gekozen wordt, de dosering en de duur van de behandeling is afhankelijk van de ernst en locatie van de bloeding, het VWD subtype, de VWF en factor VIII restactiviteit en specifieke patiënt karakteristieken.
Tranexaminezuur
Tranexaminezuur kan oraal en intraveneus toegediend worden. Bij kinderen is de dosering tranexaminezuur oraal 25-50 mg/kg per 24 uur, verdeeld over drie giften; intraveneus 25 mg/kg in 3xdd. Bij volwassenen is de dosering tranexaminezuur oraal 1g 3-4x per dag. Lokale tranexaminezuur kan toepast worden in de vorm van mondspoeling 5%: 4x/dag 2 minuten spoelen met 10ml of bijten op een gaasje gedrenkt in tranexaminezuur.
Desmopressine
DDAVP kan intraveneus (Minrin®) of subcutaan (Octostim®) toegediend worden in een dosering van 0,3 mcg/kg of als neusspray (Octostim® spray 150 microgram/dosis, 1 puff in ieder neusgat bij volwassenen en kinderen > 40 kg en 1 puff in een neusgat bij kinderen tussen de 20-40 kg als de streefspiegels daarmee gehaald worden en er geen contra-indicaties voor DDAVP zijn. Houd hierbij rekening met de tachyfylaxie van DDAVP (Mannucci, 1992).
Stollingsfactorconcentraten
Er zijn 3 groepen stollingsfactorconcentraten te onderscheiden op basis van de verhouding VWF en factor VIII:
Omdat de factor VIII productie en secretie normaal is in patiënten met VWD kan infusie van exogeen VWF, leidend tot stabilisatie en stijging van endogeen factor VIII, samen met exogeen factor VIII leiden tot ongewenst hoge factor VIII spiegels. Gebruik van stollingsfactorconcentraat met een ratio VWF/factor VIII > 10, bij patiënten met een laag factor VIII gehalte, geeft pas een stijging van het endogene factor VIII na 6-8 uur (Borel-Derlon, 2007; Mannucci, 2013). In het geval van een bloeding dient daarom bij deze patiënten gelijktijdig factor VIII toegediend te worden. Daarnaast dient rekening gehouden te worden met de half waarde tijd van de verschillende producten en de ampulgrootte (in het geval van behandeling van jonge kinderen).
Naast het verschil in VWF/factor VIII ratio verschilt ook de in vivo recovery tussen verschillende stollingsfactorconcentraten (Kessler, 2011).
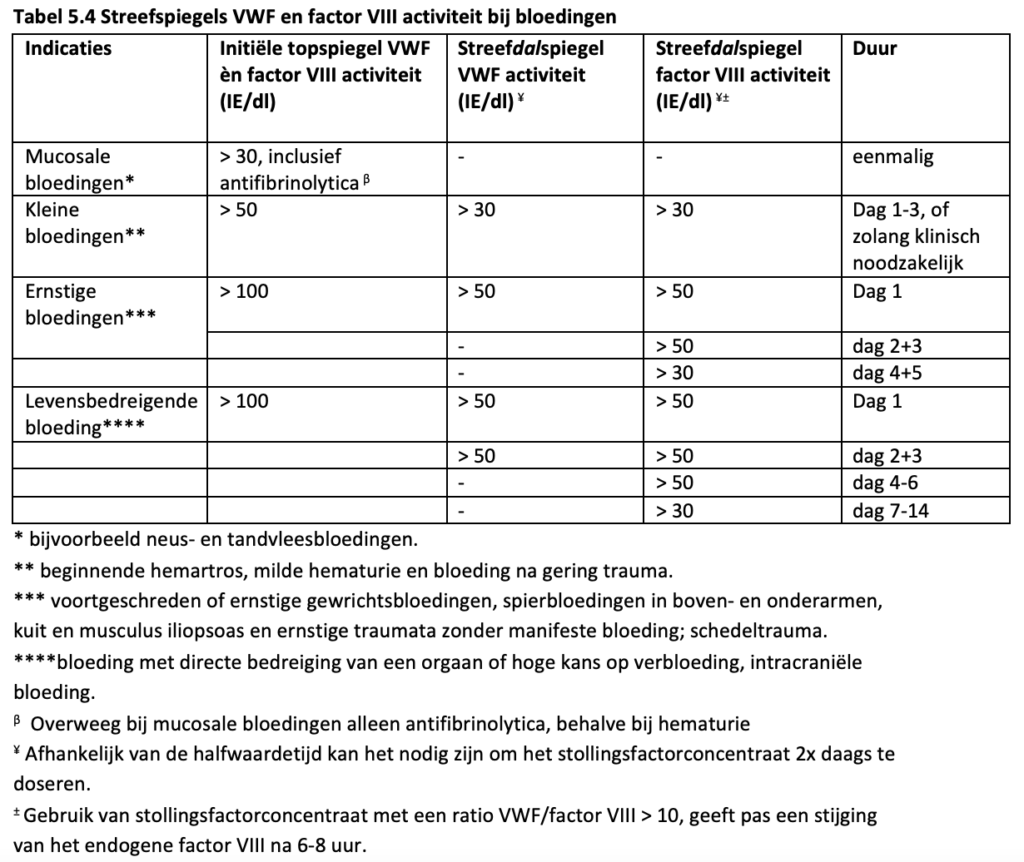
Streefspiegels VWF en factor VIII bij bloedingen
Er zijn geen gerandomiseerde studies naar de effectiviteit en veiligheid van verschillende doseringen en streefspiegels van VWF en/of factor VIII voor de behandeling van bloedingen. Het huidige advies is gebaseerd op enkele prospectieve studies en retrospectieve case series van VWD patiënten die behandeld werden voor bloedingen met VWF(/factor VIII) concentraat omdat ze niet (voldoende) responsief waren op DDAVP of er een contra-indicatie voor hadden. Daarnaast is er gebruik gemaakt van verschillende consensus richtlijnen en de bijbehorende literatuurlijsten (Nichols, 2008; NVHB, 2009; Lassila, 2011; Laffan, 2014; Connell, 2021) en enkele overzichtsartikelen (Mannucci, 2004; Leebeek, 2016).
Prospectieve studies
Mannucci et al. (Mannucci, 2002) omschreven een prospectieve multicenter studie waarin 81 VWD patiënten werden geïncludeerd. Er werden 87 bloedingen behandeld in 14 VWD patiënten (3 VWD type 1, 7 VWD type 2, 4 VWD type 3) met het VWF/factor VIII concentraat Alphanate (VWF/factor VIII ratio 1.6). Van de bloedingen waren er 73% gastro-intestinaal, 15% nasofaryngeaal, 9% muskuloskeletaal en 4% urogenitaal. In het vooraf opgestelde behandelprotocol werd een oplaaddosis gegeven van 40 IE/kg (50 IE/kg in pediatrische patiënten) VWF, met als streefspiegel een VWF van 100 IE/dL. Vervolgdoseringen werden niet gestandaardiseerd. Alle bloedingen kwamen onder controle met deze behandeling. Er was sprake van frequentere en hogere vervolgdoseringen in patiënten met VWD type 3 in vergelijking met patiënten met VWD type 2A of VWD type 1 (aantal doseringen: 3 vs. 1 vs. 1; dosering per infusie 60 vs. 40 vs. 50 IE/kg).
Gill et al. (Gill, 2003) voerden een prospectieve studie uit waarbij 33 VWD patiënten (27% VWD type 1, 24% VWD type 2, 36% VWD type 3 en 12% niet geclassificeerd), behandeld werden met Haemate-P voor 53 ernstige bloedingen. Het behandelprotocol was als volgt: een oplaaddosis van 60-80 IE/kg VWF gevolgd door een onderhoudsdosering van 40-60 IE/kg iedere 8-12 uur voor 3 dagen, eventueel gevolgd door een dagelijkse dosering van 40-60 IE/kg voor een totaal van 7 dagen. In de eerste 3 dagen was de streef VWF > 50 IE/dL. De gemiddelde oplaaddosis was 67 IE/kg, de gemiddelde onderhoudsdosering was 74 IE/kg/dag met een gemiddeld aantal infusies van 2 en een gemiddelde behandelduur van 3 dagen.
Borel-Derlon et al. (Borel-Derlon, 2007) rapporteerden een serie van 50 patiënten vanuit een prospectieve Europese en een prospectieve Franse studie die behandeld werden met Wilfactin (VWF/factor VIII ratio > 10). In 26 patiënten (waarvan 58% VWD type 3) werden 139 bloedingen behandeld. Hiervan was 32% musculoskeletaal, 38% oraal-nasofaryngeaal, 21% urogenitaal en 8% gastro-intestinaal. In de Franse studie werd er een oplaaddosis gegeven van 50 IE/kg en werd er gelijktijdig een oplaaddosis factor VIII van 30-40 IE/kg geadviseerd in patiënten met een ernstige bloeding en patiënten die een eigen factor VIII gehalte < 20 IE/dL hadden. Nadien werd er iedere 12-24 uur een dosering van 30-50 IE/kg gegeven zolang klinisch noodzakelijk was. In de Europese studie werd een oplaaddosis van 60 IE/kg gegeven. Bij spier- of gewrichtsbloedingen werd daarnaast een factor VIII concentraat geadviseerd met een streefspiegel van 80 IE/dL factor VIII. Als vervolg dosering werden 2 dagelijkse infusies van 60 IE/kg geadviseerd voor 48 uur, gevolgd door dagelijkse of om de dag toedieningen met een VWF en factor VIII streefspiegel van 30 IE/kg, zolang klinisch noodzakelijk was. De gemiddelde dosering Wilfactin was 42 IE/kg met gemiddeld 3 infusies verdeeld over 3 dagen. In 38% van de bloedingen werd tranexaminezuur voorgeschreven. Opvallend was dat 13 van de 18 ernstige bloedingen (opname noodzakelijk) ontstonden bij patiënten met een factor VIII gehalte < 20 IE/dL. Slechts de helft van deze patiënten kreeg de geadviseerde factor VIII bolus toegediend. Ook in 47% van de patiënten met een niet ernstige bloeding en een eigen factor VIII < 20% werd geen extra factor VIII concentraat toegediend. Toch werd in > 87% van deze patiënten een ‘goede’ tot ‘excellente’ klinische response op de behandeling beschreven.
Berntorp et al. (Berntorp, 2009) beschreven de effectiviteit van het VWF/factor VIII concentraat Wilate (VWF/factor VIII ratio 1) in 44 patiënten met VWD (55% met VWD type 3). Het behandelprotocol voor bloedingen bestond uit een oplaaddosis van 20-50 IE/kg. De daadwerkelijke dosering en duur van behandeling werd overgelaten aan de behandelaar. In totaal werden 1095 spontane of post-traumatische bloedingen behandeld (565 gewrichtsbloedingen, 94 epistaxis, 145 gastro-intestinale bloedingen, 34 mondbloedingen, 62 menorragie, 195 overige bloedingen). Hiervan trad 92% op in patiënten met VWD type 3. Er was een gemiddelde dosering van 29 IE/kg/dag nodig en gemiddeld 1.9 behandeldagen om de bloeding te stoppen. Opvallend was dat voor het behandelen van gastro-intestinale bloedingen meer Wilate werd gegeven (gemiddeld 44 IE/kg/dag) en dat de behandeling langer duurde (gemiddeld 4 dagen). In 96% van de bloedingen was er sprake van een ‘goede’ tot ‘ excellente’ klinische response op de behandeling.
Dunkley et al. 2010 (Dunkley, 2010) beschreven een kleine prospectieve studie van 23 VWD patiënten (7 VWD type 1, 9 VWD type 2, 7 VWD type 3) waarbij 5 patiënten werden behandeld voor 9 niet-chirurgische bloedingen (5 mucosale bloedingen en 4 niet mucosale bloedingen). Patiënten werden behandeld met een VWF/factor VIII concentraat (ratio > 1), volgens het volgende behandelprotocol: voor kleine bloedingen 1 tot 2 doses VWF van 40-50 IE/kg voor alle patiënten. Voor grote bloedingen bij patiënten met VWD type 1 werd een oplaaddosis van 40 IE/kg geadviseerd, gevolgd door 40-50 IE/kg iedere 8-12 uur met een VWF streefspiegel van 50 IE/dL voor 3 dagen, gevolgd door 40-50 IE/kg dagelijks voor een totaal van 7 dagen. Bij patiënten met VWD type 2 en 3 werd bij grote bloedingen een oplaaddosis van 50-60 IE/kg geadviseerd, gevolg door 40-60 IE/kg iedere 8-12 uur met een VWF streefspiegel van 50 IE/dL voor 3 dagen, gevolgd door 40-60 IE/kg dagelijks voor een totaal van 7 dagen. De gemiddelde dagelijkse dosering was 27 IE/kg factor VIII, met gemiddeld 2 infusies en een gemiddeld behandelduur van 2 dagen. De klinische effectiviteit van de behandeling werd beoordeeld als ‘goed’ tot ‘excellent’.
Nowak-Göttl et al. (Nowak-Göttl, 2013) rapporteerde het effect van Wilate in 15 kinderen < 6 jaar met VWD (5 VWD type 1, 4 VWD type 2, 6 VWD type 3). Het behandeladvies voor spontane en traumatische bloedingen was 20-50 IE/kg. In totaal werden 68 bloedingen (46 kleine, 18 milde en 8 ernstige bloedingen) behandeld in 11 patiënten. In 82% van de bloedingen was 1 dag behandeling voldoende. Kleine bloedingen werden behandeld met een gemiddelde dosering van 30 IE/kg, milde bloedingen met een gemiddelde dosering van 38 IE/kg en ernstige bloedingen met een gemiddelde dosering van 70 IE/kg. De klinische effectiviteit van de behandeling werd in alle bloedingen beoordeeld als ‘goed’ tot ‘excellent’.
Retrospectieve case series
Lillicrap et al. (Lillicrap, 2002) rapporteerden een serie van 97 VWD patiënten, die tussen 1991 en 1996 werden behandeld voor in totaal 344 bloedingen (32 bloedingen in patiënten met VWD type 1, 17 bloedingen in VWD type 2A, 60 bloedingen in VWD type 2B, 208 bloedingen in VWD type 3 en 27 bloedingen in ‘overige’). Ze werden behandeld met VWF/factor VIII concentraat Haemate-P (VWF/factor VIII ratio > 1). De gemiddelde dosering voor het behandelen van een bloeding in de gehele groep was 55 IE/kg. Voor patiënten met VWD type 1, 2B en 3 was dit 45-55 IE/kg en waren er 1,5-3 infusies nodig. Voor patiënten met type 2A was dit 70 IE/kg met gemiddeld 1,5 infusies. Ongeveer 35% van de patiënten hadden extra behandeling nodig na de 1e behandeldag. In 93-100% van de bloedingen was er sprake van een ‘goed’ tot ‘uitstekend’ klinisch resultaat van de behandeling.
Federici et al. (Federici, 2007) beschreef een serie van 100 VWD patiënten die behandeld werden met Haemate-P. Hiervan hadden 23 patiënten VWD type 1, 40 patiënten VWD type 2 en 37 patiënten VWD type 3. Er werden 59 patiënten (54% VWD type 3 waarvan 80% VWF < 10 IE/dL) behandeld voor in totaal 280 bloedingen (5 tandvlees, 17 menorragie, 48 hemartrose, 13 hematomen, 81 gastro-intestinaal, 22 post-traumatisch, 70 epistaxis, 18 multipele, 6 overige). Er werden geen streefspiegels benoemd. De gemiddelde dosering Haemate-P, op basis van VWF- act, was 72 IE/kg/dag. In deze groep waren 11 patiënten met een ernstige bloedingsfenotype, deze patiënten waren verantwoordelijk voor 64% van de bloedingen en 72% van het product gebruik, met een gemiddeld gebruik van 101 IE/kg/dag. Voor de behandeling van gastro-intestinale bloedingen en hemartrose werd het meeste Haemate-P gebruikt (42% resp. 21% van de totale hoeveelheid). In 95% van de patiënten was er sprake van een ‘goede’ of ‘excellente’ klinische response op de behandeling.
Federici et al. (Federici, 2010) bekeken in 2010 ook nog een groep van 120 VWD patiënten die tussen 2002 en 2006 behandeld werden met het VWF/factor VIII concentraat Alphanate of Fanhdi (VWF/factor VIII ratio 1.6). Er werden 55 patiënten (22 VWD type 1, 36 VWD type 2, 7 VWD type 3) behandeld voor 114 bloedingen. De meest voorkomende bloedingen waren hematomen (24%), epistaxis (19%) en gastro-intestinale bloedingen (17%). Er werden geen streefspiegels geadviseerd. Wel werd verwezen naar de ACIE richtlijn van de Italian Association of Hemophilia Centers uit 2002 (Federici, 2002). In deze richtlijn wordt een enkele dosis VWF(/factor VIII) concentraat van 20 IE/kg geadviseerd bij spontane of post-traumatische bloedingen met een factor VIII streefspiegel > 30 IE/dL. De gemiddelde dosering, op basis van VWF, was 40 IE/kg/dag. In 97% van de bloedingen was er sprake van een ‘goede’ tot ‘ excellente’ klinische response op de behandeling.
Khair et al. (Khair, 2015) onderzochten het gebruik van Wilate (VWF/factor VIII ratio 1) in 47 kinderen met VWD. Er werden 44 bloedingen beschreven in 15 kinderen (7 VWD type 1, 2 VWD type 2, 6 VWD type 3). De geadviseerde dosering Wilate voor de behandeling van bloedingen was 20-50 IE/kg. Van de 29 kleine bloedingen (epistaxis, menorragie, gewrichtsbloeding) was er in 86% sprake van een ‘excellente’ klinische response op een enkele dosis Wilate van gemiddeld 58 IE/kg. De 15 grote bloedingen (menorragie en intra-abdominaal) werden behandeld met een gemiddelde oplaaddosis van 51 IE/kg, waarna in 93% ten minste 1 vervolg dosering nodig was.
Overzichtsartikelen
In een review van Michiels et al. (Michiels, 2007) werd het volgende behandelprotocol beschreven: bij een ernstige bloeding: een oplaaddosis van 60-80 IE/kg VWF, gevolgd door 40 IE/kg iedere 12 uur gevolgd door 30 IE/kg iedere 24 uur met als streefspiegel een VWF > 60 IE/dL voor 4-7 dagen. Voor een mucocutane bloeding eenmalig 40-60 IE/kg VWF om een adequate VWF activiteit te krijgen > 12 uur. Voor een musculoskeletale bloeding bij patiënten met VWD type 3: een oplaaddosis van 60 IE/kg VWF gevolgd door 40-60 IE/kg/dag voor 3 dagen, om factor VIII en VWF act> 40 IE/dl te houden voor enkele dagen.
In 2004 verscheen een overzichtsartikel van Mannucci et al. (Mannucci, 2004) met het volgende behandeladvies voor spontane bloedingen bij patiënten met ernstige VWD (VWF < 10IE/dL): een dosering van 25 IE/kg (+20% bij kinderen) met als streefspiegel een factor VIII level > 30 IE/dL, totdat de bloeding stopt (meestal 2-4 dagen).
In twee overzichtsartikelen van Leebeek et al. (Leebeek, 2016; Leebeek, 2019) wordt voor kleine tot milde bloedingen een oplaaddosis van 20-40 IE/kg VWF geadviseerd met een streef VWF piekspiegel > 50-80 IE/dL op dag 1, gevolgd door een VWF dalspiegel > 30 IE/dL voor 1-3 dagen. Bij een ernstige bloeding wordt een oplaaddosis van 50 IE/kg VWF geadviseerd met een streef VWF piekspiegel > 100 IE/dL op dag 1, gevolgd een VWF dalspiegel van > 50 IE/dL voor 3-10 dagen.
In een overzichtsartikel van Lavin en Donell (Lavin, 2016) wordt bij kleine bloedingen een VWF streefspiegel > 30 IE/dL geadviseerd door middel van DDAVP of VWF concentraten met of zonder tranexaminezuur. Bij grote bloedingen wordt een VWF streefspiegel van ≈ 100 IE/dL geadviseerd door middel van VWF concentraten in combinatie met tranexaminezuur. Bij een persisterende bloeding kan een trombocytenconcentraat gegeven worden.
Castaman (Castaman, 2020) adviseert een enkele of dagelijkse dosis voor 2-3 dagen (afhankelijk van de ernst van de bloeding) van 20-60 IE/kg VWF voor spontane bloedingen in patiënten met een VWF < 10 IE/dL.
Adviezen in consensus richtlijnen
De recent gepubliceerde ASH ISTH NHF WHF 2021 VWD richtlijn (Connell, 2021) doet geen uitspraken over de behandeling van bloedingen bij de ziekte van von Willebrand.
In de Britse VWD richtlijn geschreven door de UKHCDO in 2014 (Laffan, 2014) en gebaseerd op literatuur vanaf 2002, wordt geadviseerd om kleine bloedingen te behandelen met DDAVP en tranexaminezuur. Als de patiënt niet responsief is op DDAVP of in geval van een acute of ernstige bloeding moet behandeld worden met VWF(/factor VIII) concentraat of een combinatie van een puur VWF en een puur factor VIII product. Er worden geen streefspiegels geadviseerd.
De VWD richtlijn van de Nordic Haemophilia Council (Lassila, 2011) adviseert bij mucosale bloedingen in patiënten met milde von Willebrand ziekte, oraal of lokaal tranexaminezuur. Als dat niet voldoende is, of als er sprake is van een ernstige bloeding, wordt behandeling met eenmalig DDAVP of een VWF(/factor VIII) concentraat geadviseerd, gedoseerd op basis van de VWF restactiviteit. Bij patiënten met een lage VWF wordt een dosering van 50 IE/kg VWF geadviseerd. Er worden geen streefspiegels gegeven.
In de Nederlandse richtlijn Diagnostiek en behandeling van hemofilie en aanverwante hemostasestoornissen van de NVHB uit 2009 (NVHB, 2009) worden het volgende behandelprotocol beschreven: bij milde mucosale bloedingen (epistaxis/tandvlees) een oplaaddosis Haemate-P van 20 IE/kg factor VIII, meestal eenmalig. Bij spontane of traumatische bloedingen een oplaaddosis Haemate-P van 20-40 IE/kg factor VIII, meestal eenmalig. Daarnaast wordt in de richtlijn tranexaminezuur aanbevolen bij bloedingen in weefsels met een hoge fibrinolytische activiteit, zoals de slijmvliezen. Tranexaminezuur is onvoldoende effectief bij spier- of gewrichtsbloedingen. Er worden de volgende aanbevelingen gedaan: (I) tranexaminezuur is vaak afdoende bij mucosale bloedingen of kan anders gecombineerd worden met DDAVP of VWF(/factor VIII) concentraat. (II) Tranexaminezuur is effectief bij menorragie. (III) Tranexaminezuur is gecontra-indiceerd bij hematurie in verband met risico op kolieken en obstructie. Ook wordt er geadviseerd om extreem hoge factor VIII spiegels (> 200 IE/dL) te vermijden in verband met het risico op trombose.
De Amerikaanse VWD richtlijn van het ‘National Heart, Lung, and Blood Institute (NHLBI)’ gepubliceerd in 2008 (Nichols, 2008) adviseert de volgende behandeling en streefspiegels op basis van gepubliceerde case series en consensus expert opinie: voor grote bloedingen een oplaaddosis VWF concentraat van 40-60 IE/kg, met een streefspiegel VWF en factor VIII > 100 IE/dl, gevolgd door een onderhoudsdosering iedere 8-24 uur van 20-40 IE/kg, met een VWF / factor VIII streefspiegel van > 50 IE/dL voor ten minste 7-10 dagen. Voor kleine bloedingen wordt een oplaaddosis van 30-60 IE/kg, gevolgd door een onderhoudsdosering iedere 8-24 uur geadviseerd, met een VWF / factor VIII van > 50 IE/dL voor ten minste 1-5 dagen geadviseerd. Daarnaast wordt geadviseerd om factor VIII niet boven 250-300 IE/dL en VWF niet boven 200 IE/dL te laten stijgen in verband met het risico op trombose (Makris, 2002; Mannucci, 2002).
Adviezen voor specifieke bloedingen
In een aantal richtlijnen en artikelen wordt aandacht besteed aan maatregelen bij specifieke bloedingen. Deze staan hieronder weergegeven.
Mucosale bloedingen
De VWD richtlijn van de Nordic Haemophilia Council (Lassila, 2011) adviseert voor mucosale bloedingen in patiënten met milde von Willebrand ziekte, oraal of lokaal tranexaminezuur. Als dat niet voldoende is, of als er sprake is van een ernstige bloeding, wordt behandeling met eenmalig DDAVP of een VWF(/factor VIII) concentraat geadviseerd. In de Nederlandse richtlijn Diagnostiek en behandeling van hemofilie en aanverwante hemostasestoornissen van de NVHB uit 2009 (NVHB, 2009) worden ook de volgende aan de volgende aanbevelingen gedaan ten aanzien van de behandeling van mucosale bloedingen: tranexaminezuur is vaak afdoende bij mucosale bloedingen of kan anders gecombineerd worden met DDAVP of VWF(/factor VIII) concentraat; tranexaminezuur is effectief bij menorragie; tranexaminezuur is gecontra-indiceerd bij hematurie in verband met risico op kolieken en obstructie.
Hematurie
De adviezen rondom de behandeling van hematurie bij patiënten met de ziekte van von Willebrand zijn gelijk aan de adviezen die gegeven worden bij de behandeling van hematurie bij patiënten met hemofilie (NVHB, 2020). DDAVP is gecontra-indiceerd bij hematurie vanwege het advies om bij hematurie geen vochtrestrictie aan te houden maar juist een krachtig hydratie na te streven. Gelijktijdig gebruik van DDAVP kan leiden tot een ernstige hyponatriëmie (De la Corte-Rodriguez, 2020). Antifibrinolytica bij bloedingen uit de bovenste urinewegen mogen niet toegepast worden in verband met de kans op retentie van stolsels in de ureteren en urineblaas (Mannucci, 1998) hetgeen kan resulteren in mogelijke urinewegobstructie (Pitts, 1986).
Gastro-intestinale bloedingen
Gastro-intestinale bloedingen bij patiënten met de VWD kunnen verschillende oorzaken hebben. Een belangrijke oorzaak van gastro-intestinale bloedingen is het ontstaan van angiodysplasieën ten gevolge van dysfunctionele primaire hemostase, met name bij patiënten van middelbare en oudere leeftijd (prevalentie 1,1-6,5%, bij leeftijd > 50 jaar tot 10%) (Lassila, 2011). In een internationale survey onder 4503 VWD patiënten werden angiodysplasieën met name gerapporteerd bij patiënten met VWD type 2 (2%) en VWD type 3 (4.5%), bij oudere patiënten met VWD type 2 en 3 zou dit percentage zelfs oplopen tot 11.5% (Fressinaud, 1993). Een van de hypothesen voor het ontstaan van angiodysplasieën in deze VWD patiënten is dat de afwezigheid van met name de hoge moleculair gewicht VWF multimeren, via een negatieve modulatie van VEGFR-2, leidt tot verhoogde endotheel cel proliferatie en neovascularisatie (Starke, 2011). In een overzichtsartikel van Franchini et al. (Franchini, 2014) wordt een overzicht gegeven van de verschillende behandelopties. Voor behandeling van acute bloedingen wordt VWF(/factor VIII) concentraat (40-60 IE/dag) in combinatie met tranexaminezuur geadviseerd, maar ook zijn lokale invasieve behandelingen zoals endoscopische thermocoagulatie en operatief ingrijpen soms noodzakelijk. Ook behandeling met recombinant FVIIa (Meijer, 2001) en trombocytenconcentraat (Castillo, 1991) is beschreven. Opvallend is dat er gemiddeld meer VWF(/factor VIII) concentraat nodig is voor de behandeling van gastro-intestinale bloedingen dan voor andere acute bloedingen (Federici, 2007; Berntorp, 2009). Secundaire profylaxe is soms noodzakelijk om recidiverende gastro-intestinale bloedingen te voorkomen, maar ook dit lijkt minder effectief dan bij bijvoorbeeld recidiverende gewrichtsbloedingen. Een dosering VWF van 40-60 IE/kg, 2-3x per week leidde in een studie van Abshire et al. (Abshire, 2013) tot een daling van de jaarlijkse annual gastro-intestinal bleeding rate van 8,4 naar 6, ten opzichte van een daling van 15,6 naar 1,3 bij gewrichtsbloedingen. Een suggestie die gegeven wordt in de literatuur is het geven van Vonicog alfa, een puur recombinant VWF product, bij behandeling van gastro-intestinale bloedingen, omdat dit middel ultra grote VWF multimeren bevat (Leebeek, 2017; Mannucci, 2019). Enkele case reports beschrijven succesvolle behandeling van patiënten met de angiogenese remmer thalidomide (50-100mg per dag) of atorvastatine (tot 80 mg per dag) (Nomikou, 2009; Alikhan, 2010). In 2014 werd ook een retrospectieve studie gepubliceerd waarin gekeken werd naar het natuurlijk beloop van angiodysplasieën in VWD. Patiënten met congenitale VWD werden geïncludeerd als ze tenminste één episode van gastro-intestinaal bloedverlies hadden waarbij er geen duidelijke oorzaak te vinden was, of wanneer dit te gevolge van angiodysplasieën was. Er werden 48 patiënten geïncludeerd, waarvan 37,5% patiënten met VWD type 3, 22,9% patiënten met VWD type 2A maar ook 18,7% met VWD type 1. Acute bloedingen werden in 95% van de patiënten succesvol behandeld. Profylaxe was in 85% van de patiënten effectief in het voorkomen van recidieven. Thalidomide was niet succesvol in de 5 patiënten waarin dit werd voorgeschreven.
Platelet type VWD
Bij platelet type VWD is er sprake van een afwijkend GPIb op de bloedplaatjes, waardoor er verhoogde affiniteit voor VWF ontstaat. Door deze spontane binding aan GPIb is er bij platelet type VWD vaak een trombopenie, die nog verder kan toenemen bij het geven van DDAVP of VWF/factor VIII concentraat. In 2020 publiceerde de Platelet Physiology Subcommittee van de ISTH een richtlijn voor de diagnostiek en management van platelet type VWD (Othman, 2020). Voor de behandeling van een grote bloeding wordt VWF concentraat in combinatie met 1 HLA gematched trombocytenconcentraat geadviseerd. Als er sprake is van een refractaire bloeding kan recombinant factor VII overwogen worden in een dosering van 90 microgram/kg, iedere 2 uur indien nodig. Kleine bloedingen kunnen behandeld worden met tranexaminezuur 10-30 mg/kg iedere 8 uur, en eventueel DDAVP, als hier geen contra-indicaties voor zijn en het niet leidt tot toename van de trombopenie.
Tromboserisico
In meerdere richtlijnen wordt geadviseerd om extreem hoge factor VIII spiegels te vermijden (> 150-300 IE/dL). In de studie van Koster et al. (Koster, 1995) bleek een factor VIII waarde > 150 IE/dL een onafhankelijke risicofactor te zijn voor het ontwikkelen van een diep veneuze trombose met een odds ratio (OR) van 4,8 (95%CI: 2,3-10,0). Een systematische review uit 2012 (Coppola, 2012) bekeek trombotische complicaties gerelateerd aan factor substitutie in patiënten met hemofilie en VWD in prospectieve studies over een periode van 20 jaar, en het bleek dat tromboembolische complicaties zeldzaam waren en voornamelijk oppervlakkige tromboflebitis betroffen.
Referenties
Bewijskracht literatuur
Level 2-3, consistent
Zoeken en selecteren
Er werd voor de algemene uitgangsvraag geen systematisch literatuuronderzoek verricht. Er werd voor deze uitgangsvraag gebruik gemaakt van de in 2021 door de American Society of Hematology (ASH), International Society on Thrombosis and Haemostasis (ISTH), National Hemophilia Foundation (NHF), and World Federation of Hemophilia (WFH) opgestelde en gepubliceerde evidence-based richtlijn en de bijbehorende referentielijst; de Britse richtlijn voor diagnostiek en management van VWD van de United Kindom Haemophilia Centre Doctors Organisation (UKHCDO) uit 2014; de Nordic Haemophilia Council’s guideline uit 2011; de richtlijn Diagnostiek en behandeling van hemofilie en aanverwante hemostasestoornissen van de Nederland Vereniging van Hemofiliebehandelaars (NVHB) uit 2009 en de VWD richtlijn van de Amerikaanse National Heart, Lung, and Blood Institute (NHLBI) uit 2008.
Voor advies rondom specifieke bloedingen werd het volgende systemische literatuuronderzoek verricht:
Gastro-intestinale bloedingen
Er werd voor deze vraag een systematische review verricht met de volgende zoektermen: “willebrand” and “angiodysplasia” not “acquired” . Dit resulteerde in 104 publicaties. Na titel en abstract screening resulteerde dat in 28 relevante publicaties, waaronder veel case reports en reviews.
Zoekverantwoording
De zoekverantwoording is opgenomen in diverse richtlijnen en bijbehorende (online) supplementen (Nichols, 2008; NVHB, 2009; Lassila, 2011; Laffan, 2014; Connell, 2021)
Evidence tabellen
De evidence tabellen van de gebruikte richtlijnen zijn weergegeven in het EtD raamwerk van de ASH ISTH NHF WFH richtlijn (Connell, 2021), online beschikbaar; op de website van de British Committee for Standards in Haematology (Laffan, 2014); op de website van de Nordic Haemophilia Counsil (Lassila, 2011); op de website van de NHLBI richtlijn (Nichols, 2008).
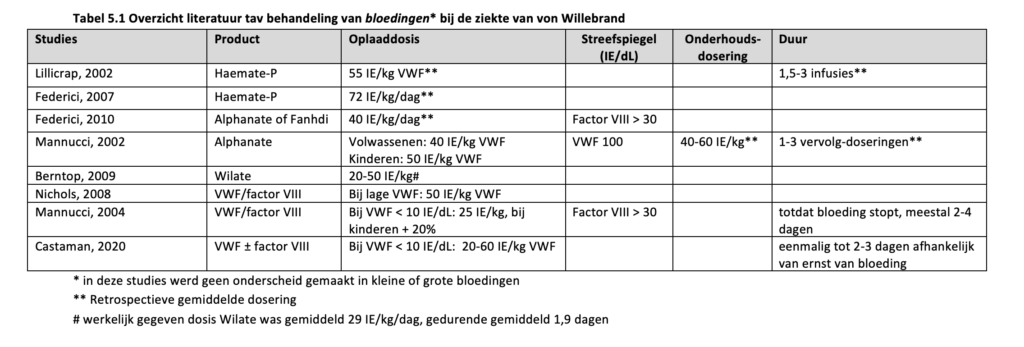
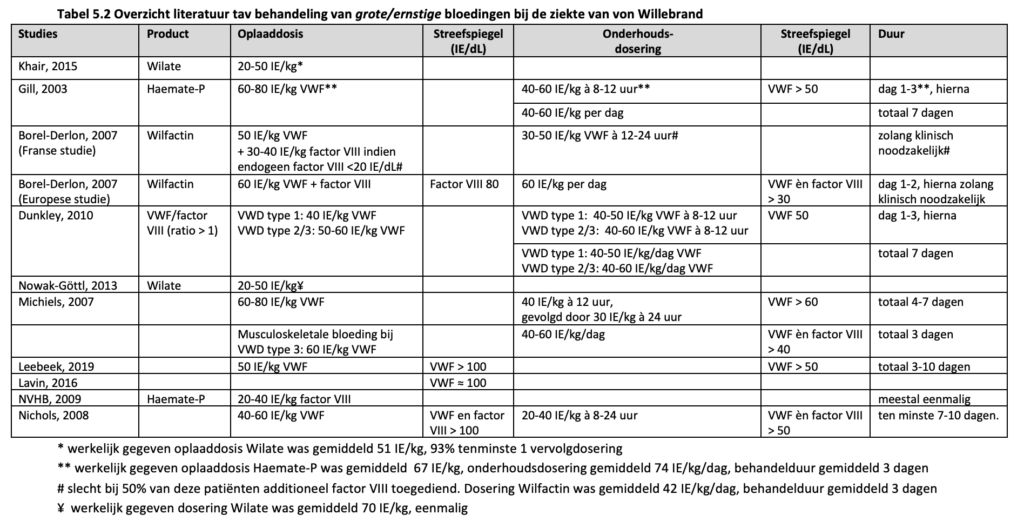
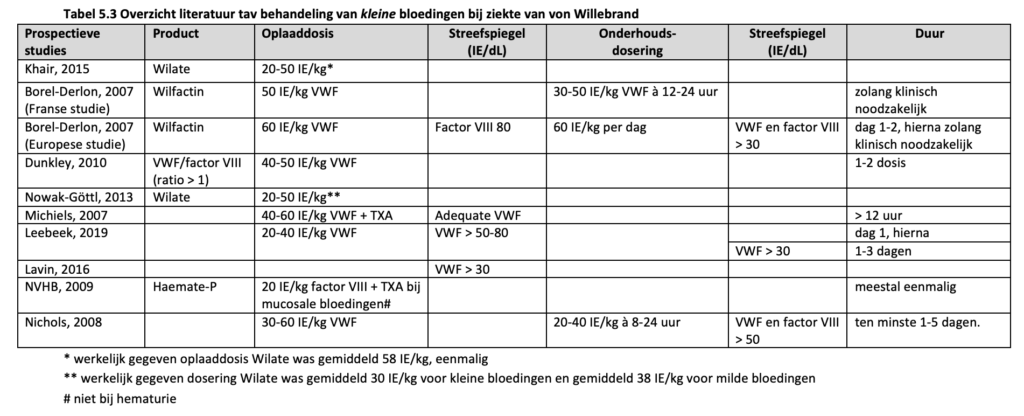
De in de studies gerapporteerde streefspiegels voor VWF en factor VIII bij de behandeling van een bloeding bij VWD zijn weergegeven in de tabellen 5.1 (geen onderscheid in grote/kleine bloedingen), 5.2 (grote bloedingen) en 5.3 (kleine bloedingen).
Overwegingen
Streefspiegels VWF en factor VIII bij bloedingen
Er is veel variatie in de behandeling van bloedingen en de streefspiegels voor VWF en factor VIII in studies, reviews en richtlijnen. In de verschillende consensus richtlijnen wordt een onderscheidt gemaakt in grote en kleine bloedingen. Daarnaast worden er ook adviezen gegeven voor specifieke bloedingen. Over het algemeen wordt een bloeding behandeld door het geven van een oplaaddosis VWF(/factor VIII) concentraat. De dosering hiervan varieert bij kleine bloedingen tussen de 20-60 IE/kg VWF/factor VIII, bij grote bloedingen varieert deze dosering tussen de 20-70 IE/kg VWF/factor VIII, waarbij er meestal gekozen wordt voor een dosering van ongeveer 50 IE/kg. Ook het beleid rondom vervolgdoseringen is variabel. Bij kleine bloedingen wisselt dit van een eenmalige dosis tot 5 dagen behandeling en bij grote bloedingen varieert dit van 3 tot 7 dagen behandeling, afhankelijk van het klinisch beloop. De gerapporteerde streefspiegels VWF en factor VIII bij de behandeling van een bloeding variëren voor kleine bloedingen tussen > 30-80 IE/dL VWF en/of factor VIII na de oplaaddosis, tot > 30-50 IE/dL voor de dagen daarna. Voor grote bloedingen is dit > 80-100 IE/dL VWF en/of factor VIII na de oplaaddosis en > 30-50 IE/dL voor de dagen daarna (tabellen 5.1-5.3). In de studies werd de klinische effectiviteit van deze behandelingen voor het overgrote deel beoordeeld als ‘goed’ tot ‘excellent’.
Omdat het geven van een ‘oplaaddosis’ afhankelijk is van de nog aanwezige eigen restactiviteit VWF en factor VIII en het concentraat dat gegeven wordt, heeft het de voorkeur dat op basis van de restactiviteit van VWF en factor VIII van de patiënt berekend wordt hoeveel stollingsfactorconcentraat nodig is om een bepaalde streefwaarde te bereiken. De hoeveelheid stollingsfactorconcentraat kan berekend worden op basis van zowel de VWF concentratie en de factor VIII concentratie. Hierbij kan gebruik gemaakt worden van de onderstaande berekeningen.
Doordat de diverse VWF/factor VIII concentraten een verschillende verhouding VWF en factor VIII bevatten, dient er bij het doseren rekening te worden gehouden met wat de te verwachten stijging van zowel VWF activiteit als factor VIIII activiteit zal zijn op basis van de toegediende aantal eenheden per factor. Daarnaast dient men zich goed te realiseren dat het gebruik van een stollingsfactorconcentraat met een ratio VWF/factor VIII > 10, bij patiënten met een laag factor VIII gehalte, pas een stijging van het endogene factor VIII geeft na 6-8 uur. In het geval van een bloeding dient daarom bij deze patiënten gelijktijdig factor VIII toegediend te worden.
Adviezen voor specifieke bloedingen
Bij mucosale bloedingen kan in het geval van een milde bloeding of bij patiënten met milde VWD in eerste instantie behandeling met alleen tranexaminezuur overwogen worden. Er zijn geen studies die gerandomiseerd uitgezocht hebben, maar dit is in overeenkomst met de aanbeveling om alleen tranexaminezuur te geven bij patiënten met type 1 VWD met spiegels > 30IE/dL en een mild bloedingsfenotype die kleine mucosale ingrepen moeten ondergaan (Walsh, 1971; Forbes, 1972).
Bij hematurie is tranexaminezuur gecontra-indiceerd, net als DDAVP in verband met het risico op ernstige hyponatriëmie (De la Corte-Rodriguez, 2020) en de kans op retentie van stolsels in de ureteren en urineblaas met het risico op urinewegobstructie (Mannuci, 1998, Pitts, 1986). Gastro-intestinaal bloedverlies uit angiodysplasieën lijkt met name voor te komen bij patiënten met VWD type 2 en 3, maar komt ook voor bij VWD type 1. Hoewel behandeling van een acute bloeding met VWF/factor VIII concentraat en tranexaminezuur in de meeste patiënten werkt, is deze behandeling minder effectief dan bij andere acute bloedingen. Om recidiverende gastro-intestinale bloedingen te voorkomen is het soms noodzakelijk om profylaxe te starten. Ook hier is het effect kleiner dan het effect van profylaxe op bijvoorbeeld recidiverende gewrichtsbloedingen. Mogelijk dat het gebruik van een puur recombinant VWF product, waarbij de ultra grote VWD multimeren aanwezig zijn, effectiever is in de behandeling van acute bloedingen en het voorkomen van recidieven. Andere behandelopties zoals thalidomide of atorvastatine zijn beschreven, met wisselende resultaten.
Hoe is de behandeling rondom een ingreep of operatie?
Aanbevelingen
Onderbouwing
Inleiding
Operaties en ingrepen bij patiënten met VWD moeten, afhankelijk van de restactiviteit van VWF, onder stollingsfactorcorrectie plaatsvinden om bloedingscomplicaties te voorkomen. Er kan gebruik gemaakt worden van VWF(/factor VIII) concentraten (plasma-derived of recombinant), of van een synthetisch preparaat, DDAVP (1-deamino-8-D-arginine-vasopressine: Minrin, intraveneus; Octostim, neusspray/subcutaan), dat tijdelijk de plasmaconcentratie van VWF/factor VIII verhoogt door een release van VWF uit de endotheelcellen te bewerkstelligen. Daarnaast wordt gebruik gemaakt van antifibrinolytica (tranexaminezuur, Cyklokapron). De keuze van het preparaat is afhankelijk van het type VWD en de ingreep en patiënt afhankelijke factoren (zoals respons op DDAVP, bloedingsneiging en eventuele contra-indicaties). Het is voor iedere ingreep belangrijk dat maximale lokale hemostase wordt nagestreefd.
Conclusies
|
SORT Grade |
Conclusie |
|
C |
Hoewel gedacht wordt dat factor VIII cruciaal is in het voorkomen van chirurgische bloedingen, zal het alleen suppleren van factor VIII concentraat met name in patiënten met type 2 en type 3 VWD niet leiden tot adequate hemostase tijdens operaties. (Castaman, 2011; Connell, 2021) |
|
C |
Lokale beschikbaarheid van snelle bepalingen van VWF en factor VIII is noodzakelijk voor goede perioperatieve begeleiding van VWD patiënten. (Connell, 2021) |
|
C |
Het doseren van het stollingsfactorconcentraat Haemate-P (factor VIII:VWF 1:2,4) op basis van alleen factor VIII leidt voor VWF in meer dan 50% en voor factor VIII in meer dan 70% tot een stijging van ≥ 20 IE/dL boven de streefspiegel. (Hazendonk, 2018) |
|
C |
Het bevorderen van lokale hemostase tijdens een ingreep met gelatinesponzen, fibrinelijm en/of lokale tranexaminezuur applicatie is zinvol. (Federici, 2000; De Padua, 2020) |
|
C |
Het gebruik van alleen factorconcentraten of DDAVP met een VWF streefspiegel van > 50 IE/dL, is bij kleine ingrepen geassocieerd met een significant hoger risico op postoperatieve bloeding in vergelijking met de combinatie tranexaminezuur met een van beide behandelingen (n=59; RR, 6,29; 95% CI: 2,12-18,65). (Walsh, 1971; Forbes, 1972; Connell, 2021) |
Samenvatting literatuur
Resultaten
Algemeen
De perioperatieve behandeling van VWD is ingewikkeld, als gevolg van de verschillende typen VWD, de inter-patiënt variatie in residuaal endogeen VWF, VWF secretie en klaring, en de verschillende stollingsfactorconcentraten met wisselende VWF/factor VIII concentraties. Er zijn geen gerandomiseerde studies verricht naar de effectiviteit en veiligheid van verschillende doseringen en streefspiegels van VWF/factor VIII voor de verschillende ingrepen bij patiënten met VWD. De data die deze aanbevelingen ondersteunen komen voort uit klinische ervaring, consensus van expert opinion en observationele studies. Er wordt onderscheid gemaakt tussen kleine en grote ingrepen waarbij met name de minimale behandelduur varieert. De productkeuze, dosering en duur van behandeling is afhankelijk van het klinische scenario (type ingreep, bloedingsrisico), VWD subtype en VWF en factor VIII gehalte van de patiënt en de voorkeur van een HBC.
Tranexaminezuur
Tranexaminezuur kan oraal en intraveneus toegediend worden. Bij kinderen is de dosering tranexaminezuur oraal 25-50 mg/kg per 24 uur, verdeeld over drie giften; intraveneus 25 mg/kg in 3xdd. Bij volwassenen is de dosering tranexaminezuur 1g 3-4x per dag. Lokale tranexaminezuur kan toepast worden in de vorm van mondspoeling 5%: 4x/dag 2 minuten spoelen met 10ml of bijten op een gaasje gedrenkt in tranexaminezuur.
Desmopressine
DDAVP kan intraveneus (Minrin®) of subcutaan (Octostim®) toegediend worden in een dosering van 0,3 mcg/kg of als neusspray (Octostim® spray 150 microgram/dosis, 1 puff in ieder neusgat bij volwassenen en kinderen > 40 kg en 1 puff in een neusgat bij kinderen tussen de 20-40 kg), minimaal 30 minuten voor de ingreep, als de streefspiegels daarmee gehaald worden en er geen contra-indicaties voor DDAVP zijn. Houd hierbij rekening met de tachyfylaxie van DDAVP (Mannucci, 1992).
Stollingsfactorconcentraten
Er zijn 3 groepen stollingsfactorconcentraten te onderscheiden op basis van de verhouding VWF en factor VIII:
Omdat de factor VIII productie en secretie normaal is in patiënten met VWD kan infusie van exogeen VWF, leidend tot stabilisatie en stijging van endogeen factor VIII, samen met exogeen factor VIII leiden tot ongewenst hoge factor VIII spiegels. Gebruik van een stollingsfactorconcentraat met een ratio VWF/factor VIII > 10, bij patiënten met een laag factor VIII gehalte, geeft pas een stijging van het endogene factor VIII na 6-8 uur (Borel-Derlon, 2007; Mannucci, 2013) en moet daarom 6-8 uur voor de ingreep toegediend worden. Er kan ook gekozen worden om bij de eerste gift, gelijktijdig factor VIII toe te dienen. Daarnaast dient rekening gehouden te worden met de halfwaarde tijd van de verschillende producten en de ampulgrootte (in het geval van behandeling van jonge kinderen).
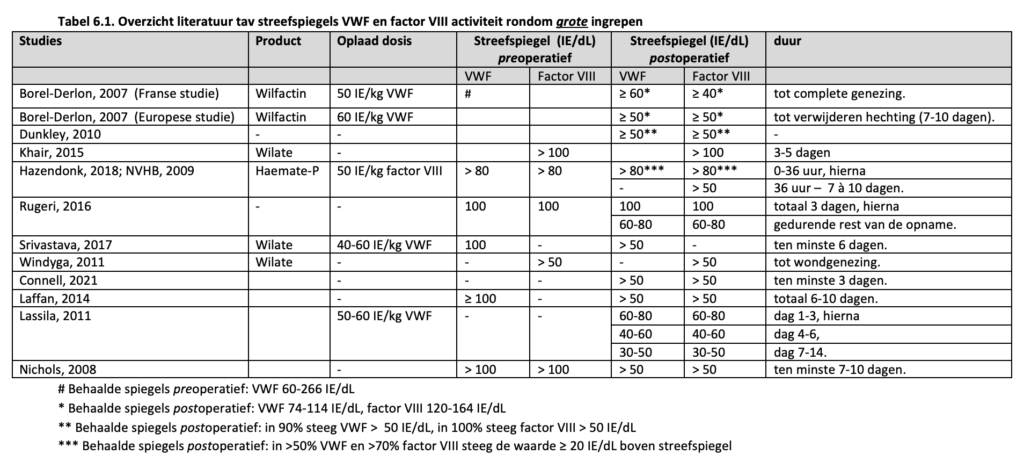
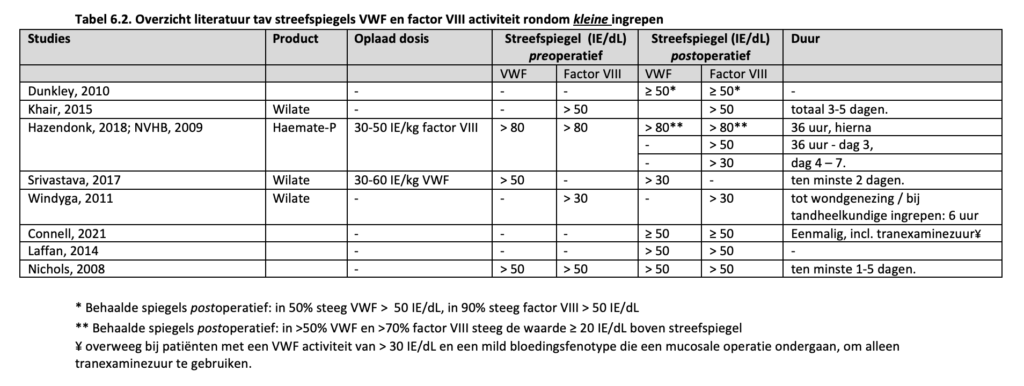
Streefspiegels VWF en factor VIII bij grote en kleine ingrepen
Er zijn geen studies naar de effectiviteit en veiligheid van verschillende doseringen en streefspiegels van VWF en/of factor VIII voor ingrepen. Het huidige advies is gebaseerd op enkele prospectieve studies en retrospectieve case series van VWD patiënten die verschillende grote operaties ondergingen, waarbij zowel de behaalde factor VIII als VWF spiegels ≥ 3 dagen postoperatief gerapporteerd werden (Connell, 2021). Daarnaast is er gebruik gemaakt van verschillende consensus richtlijnen (Nichols, 2008; NVHB, 2009; Lassila, 2011; Laffan, 2014).
Prospectieve studies
Borel-Derlon et al. (Borel-Derlon, 2007) rapporteerden een serie van 50 patiënten vanuit een Europese en een Franse studie (5 VWD type 1, 27 type 2 en 18 type 3; 72% met VWF < 10 IE/dL). Deze patiënten ondergingen in totaal 65 grote operaties (14 orthopedische, 10 gynaecologische, 7 algemene, 14 tandheelkundige en 20 gastro-intestinale ingrepen) en 43 invasieve procedures (o.a. endoscopieën, hysteroscopieën, coronair angiografie, intramusculaire en intra-articulaire injecties). Ze werden behandeld met Wilfactin (VWF/factor VIII ratio > 10). Er werd een preoperatieve oplaaddosis gegeven van 50 IE/kg VWF in de Franse studie en van 60 IE/kg VWF in de Europese studie. Bij een endogeen factor VIII gehalte van < 60 IE/dL werd er ook een bolus 12 uur voorafgaand aan de ingreep gegeven. Er werden geen preoperatieve streefspiegels vastgesteld. Postoperatieve streefspiegels waren een VWF spiegel van ten minste 60 IE/dL en een factor VIII spiegel van ten minste 40 IE/dL, tot complete genezing was bereikt in de Franse studie, of een VWF en factor VIII spiegel van ten minste 50 IE/dL tot verwijderen van de hechtingen na 7-10 dagen in de Europese studie. Preoperatieve VWF spiegels varieerden van 60-266 IE/dL. De behaalde postoperatieve VWF spiegels varieerden van 74-114 IE/dL en de factor VIII spiegels varieerden van 120-164 IE/dL. Peri-operatieve hemostase werd beoordeeld als ‘uitstekend’ in 54 operaties en ‘goed’ in 11 operaties. Er waren geen abnormale bloedingen na de invasieve procedures.
Dunkley et al. (Dunkley, 2010) beschreven een serie van 23 VWD patiënten (7 VWD type 1, 9 type 2, 7 type 3) waarbij 10 grote ingrepen en 19 kleine ingrepen plaatsvonden. Patiënten werden behandeld met een VWF/factor VIII concentraat (ratio > 1). Er werden geen specifieke pre- of postoperatieve streefspiegels gegeven, maar achteraf gekeken of de waarden binnen de normaal waarden vielen (VWF en factor VIII beide 50-200 IE/dL). De gemiddelde dosering (gebaseerd op factor VIII) voor grote ingrepen was 73 IE/kg en voor kleine ingrepen 35 IE/kg. Bij grote ingrepen werd bij 90% de streefspiegel van VWF > 50 IE/dL behaald, bij kleine ingrepen was dit 50% (voor factor VIII was dit 100% resp. 90%). De mediane factor VIII spiegel was 115 IE/dL, de mediane VWF spiegel was 85 IE/dL. Peri-operatieve hemostase werd beoordeeld als ‘uitstekend’ en ‘goed’ voor alle ingrepen. In twee patiënten steeg de factor VIII spiegel boven de 200 IE/dL. Er waren geen trombo-embolische complicaties.
Retrospectieve case series
Khair et al. (Khair, 2015) onderzochten het gebruik van Wilate (VWF/factor VIII ratio 1) in 47 kinderen met VWD (7 VWD type 1, 9 type 2, 7 type 3). Er werden 41 operaties uitgevoerd in 34 kinderen: 9 kleine ingrepen, 7 tandheelkundige en 25 grote ingrepen. De streefspiegel was een factor VIII > 50 IE/dL voor kleine ingrepen en > 100 IE/dL voor grote ingrepen, voor 3-5 dagen. De gemiddelde dosering Wilate was eenmalig 70 IE/kg voor kleine ingrepen, eenmalig 45 IE/kg voor de tandheelkundige ingrepen en gemiddeld 4 giften van 43 IE/kg voor grote ingrepen. Peri-operatieve hemostase werd beoordeeld als ‘goed’ of ‘uitstekend’ in 94% van de ingrepen. De gemiddelde maximale factor VIII spiegels waren 135 IE/dL, dit was 92 IE/dL voor VWF. Er waren geen adverse events.
Hazendonk et al. (Hazendonk, 2018) analyseerden het perioperatieve beleid met VWF/factor VIII concentraat Haemate-P (VWF/factor VIII ratio > 1) van patiënten met VWD die tussen 2000 en 2015 geopereerd werden. In totaal werden 103 patiënten met VWD (54 VWD type 1, 43 VWD type 2, 6 VWD type 3) geïncludeerd. Er werden 148 operaties uitgevoerd, waarbij er sprake was van 38 kleine ingrepen en 110 grote ingrepen. Hierbij werd de Nederlandse richtlijn gevolgd waarbij preoperatief tot 36 uur postoperatief een VWF en factor VIII streefspiegel van > 80 IE/dL werd aangehouden, met daarna voor grote ingrepen een factor VIII spiegel van > 50 IE/dL voor 7-10 dagen en bij kleine ingrepen een factor VIII > 50 IE/dL voor 3 dagen, met daarna een factor VIII > 30 IE/dL voor 4-7 dagen. Haemate-P werd gedoseerd op basis van factor VIII. Er werd een preoperatieve oplaaddosering gegeven van 50 IE/kg factor VIII voor grote ingrepen en 30-50 IE/kg factor VIII voor kleine ingrepen. In meer dan de 50% van de patiënten steeg de VWF tot ≥ 20 IE/dL boven de vastgestelde streefspiegel en in meer dan 70% was dit het geval voor factor VIII. Ook werd er stapeling van factor VIII gezien na herhaalde doseringen Haemate-P. In 8% van de patiënten steeg de factor VIII > 270 IE/dL. Er werden geen bloedingscomplicaties en geen trombo-embolische complicaties gezien.
Rugeri et al. (Rugeri, 2016) evalueerden het beloop van 12 VWD patiënten (6 type 1 VWD, 6 type 2 VWD) die in totaal 19 grote operaties ondergingen (12 totale knie protheses, 7 totale heup protheses). De streefspiegels voor VWF en factor VIII waren preoperatief tot 3 dagen postoperatief 100 IE/dL en daarna 60-80 IE/dL gedurende de rest van de ziekenhuisopname. Vier patiënten werden behandeld met DDAVP waarbij de VWF steeg naar gemiddeld 196 IE/dL. De overige 8 patiënten werden behandeld met VWF concentraten met een gemiddelde dosering van 52 IE/kg resulterend in een stijging van VWF naar gemiddeld 161 IE/dL en factor VIII naar gemiddeld 174 IE/dL. Hemostatische controle was ’uitstekend’ in 74% en ‘goed’ in 11% van de procedures. 1 VWD patiënt voldeed aan de criteria voor major bleeding, bij een adequate VWF spiegel.
Srivastava et al. (Srivastava, 2017) beschreven 28 patiënten (21 VWD type 3) die 30 ingrepen (21 grote, 9 kleine) ondergingen met Wilate (VWF/factor VIII ratio 1). Er werd een oplaaddosis gegeven van 40-60 IE/kg VWF bij grote ingrepen en 30-60 IE/kg bij kleine ingrepen. Voor grote ingrepen was de preoperatieve streefspiegel voor VWF 100 IE/dL en postoperatief > 50 IE/dL voor ten minste 6 dagen. Voor kleine ingrepen was de VWF streefspiegel preoperatief > 50 IE/dL en postoperatief > 30 IE/dL voor ten minste 2 dagen. Perioperatieve hemostase werd beoordeeld als ‘succesvol’ in 100% van de kleine ingrepen en in 95% van de grote ingrepen. De gemiddelde VWF spiegel was 41 IE/dL, voor factor VIII was dit 92 IE/dL. In 6 patiënten steeg factor VIII tot > 250 IE/dL. Er waren geen trombo-embolische complicaties.
Windyga et al. (Windyga, 2011) beschreven de effectiviteit van het VWF/factor VIII concentraat Wilate (VWF/factor VIII ratio 1) in de ‘European Wilate study’ bij 57 operaties (27 grote en 30 kleine) in 32 VWD patiënten (19 type 3 VWD, 9 type 2 VWD, 4 type 1 VWD). De streefspiegel bij grote ingrepen was een factor VIII > 50 IE/dL, preoperatief tot complete wondgenezing; bij kleine ingrepen een factor VIII > 30 IE/dL, preoperatief tot complete wondgenezing, of een totaal van 6 uur bij tandheelkundige ingrepen. De gemiddelde oplaaddosering voor grote ingrepen was 49 IE/kg, voor kleine ingrepen was dit 39 IE/kg. Voor grote ingrepen was de gemiddelde onderhoudsdosering 21 IE/kg, met gemiddeld 11 infusies in totaal. Voor kleine ingrepen was dit 22 IE/kg met gemiddeld 1,5 infusies in totaal. Er waren meer patiënten met VWD type 3 die een kleine ingreep ondergingen. De hemostatische controle werd beoordeeld als ‘uitstekend’ of ‘goed’ in 96% van de ingrepen.
Adviezen in consensus richtlijnen
Op basis van bovenstaande literatuur wordt in de ASH ISTH NHF WHF 2021 VWD richtlijn (Connell, 2021) gesuggereerd om, indien mogelijk, bij grote ingrepen een streefspiegel van zowel VWF als factor VIII ≥ 50 IE/dL aan te houden gedurende ten minste drie dagen. Het gebruik van alleen een factor VIII streefspiegel wordt afgeraden. Specifieke streefspiegels en duur van behandeling moeten individueel vastgesteld worden op basis van patiëntkarakteristieken, bloedingsfenotype, type ingreep, en laboratorium mogelijkheden. Voor kleine ingrepen wordt gesuggereerd om tranexaminezuur toe te voegen, naast het eenmalig verhogen van de VWF ≥ 50 IE/dL. Het verhogen van VWF ≥ 50 IE/dL zonder de toevoeging van tranexaminezuur wordt afgeraden. Daarnaast suggereren ze bij patiënten met een VWF activiteit van > 30 IE/dL en een mild bloedingsfenotype die een mucosale operatie ondergaan, om alleen tranexaminezuur te gebruiken. Bij tandheelkundige ingrepen moeten producten die lokale hemostase bevorderen overwogen worden. In deze richtlijn wordt ook aangegeven dat het wenselijk is om het langdurig stijgen van zowel VWF als factor VIII > 150 IE/dL en langdurig gebruik van tranexaminezuur te voorkomen. Er wordt geen advies gegeven over preoperatieve streefspiegels.
De Britse VWD richtlijn geschreven door de UKHCDO in 2014 (Laffan, 2014) en gebaseerd op literatuur vanaf 2002, adviseert voor grote ingrepen een preoperatieve factor VIII streefspiegel van ≥ 100 IE/dL, gevolgd door een factor VIII > 50 IE/dL tot 7-10 dagen postoperatief. Voor de VWF adviseren ze een spiegel van > 50 IE/dL perioperatief. Bij kleine ingrepen is een VWF en factor VIII van > 50 IE/dL voldoende.
De VWD richtlijn van de Nordic Haemophilia Council (Lassila, 2011) adviseert voor grote ingrepen een oplaad dosis van 50-60 IE/kg VWF, gevolgd door een onderhoudsdosering van 25-40 IE/kg VWF iedere 12-24 uur. De geadviseerde postoperatieve streefspiegels zijn een VWF en factor VIII van 60-80 IE/dL voor dag 1-3, 40-60 IE/dL voor dag 4-6 en 30-50 IE/dL voor dag 7-14. Hoge factor VIII spiegels (> 150 IE/dL) moeten voorkomen worden. Kleine ingrepen kunnen onder DDAVP verricht worden. Tranexaminezuur 15-20 mg/kg kan toegevoegd worden, zeker bij mucosale ingrepen.
In de Nederlandse richtlijn Diagnostiek en behandeling van hemofilie en aanverwante hemostasestoornissen van de NVHB uit 2009 (NVHB, 2009) worden het volgende advies gegeven: een preoperatieve oplaad dosis van 50 IE/kg factor VIII voor grote ingrepen en 30-50 IE/kg factor VIII voor kleine ingrepen. Daarna een dosering van 25 IE/kg tweemaal daags bij grote ingrepen en 15-25 IE/kg tweemaal daags bij kleine ingrepen, op geleide van de factor VIII concentratie. Er worden de volgende streefspiegels geadviseerd: preoperatief en de eerste 36 uur postoperatief een factor VIII en een VWF > 80 IE/dL. Bij grote ingrepen wordt daarna een factor VIII > 50 IE/dL aangehouden gedurende 7-10 dagen. Bij kleine ingrepen is dit een factor VIII > 50 IE/dL gedurende 3 dagen, gevolgd door een factor VIII > 30 IE/dL gedurende 4-7 dagen. Ook wordt er geadviseerd om extreem hoge factor VIII spiegels (> 200 IE/dL) te vermijden. Dit beleid is in de eerder beschreven studie van Hazendonk et al. geëvalueerd (Hazendonk, 2018).
De Amerikaanse VWD richtlijn van het ‘National Heart, Lung, and Blood Institute (NHLBI)’ gepubliceerd in 2008 (Nichols, 2008) adviseert de volgende streefspiegels op basis van gepubliceerde case series en consensus expert opinie: voor grote ingrepen een preoperatieve VWF / factor VIII spiegel van > 100 IE/dL, met nadien voor beide een streefspiegel van > 50 IE/dL voor ten minste 7-10 dagen. Voor kleine ingrepen wordt preoperatief een VWF / factor VIII van ten minste > 50 IE/dL voor ten minste 1-5 dagen geadviseerd. Daarnaast wordt geadviseerd om factor VIII niet boven 250-300 IE/dL en VWF niet boven 200 IE/dL te laten stijgen in verband met het risico op trombose (Makris, 2002; Mannucci, 2002).
Tranexaminezuur bij kleine ingrepen
Er zijn 8 case series beschreven waarin bij kleine ingrepen alleen suppletie therapie werd gegeven, zonder tranexaminezuur en 4 case series waarin patiënten alleen tranexaminezuur kregen bij het ondergaan van kleine ingrepen (tandextracties, circumcisies, leverbiopten, endoscopie met biopten) (Connell, 2021). In de case series met alleen suppletie therapie (streefspiegel van VWF > 50 IE/dL) zonder additioneel tranexaminezuur werd in 11% van de 281 ingrepen (95% CI, 6-19%) (Franchi, 1995; Venkataramani, 2000; Shin, 2005; Viswabandya, 2008; Rodriguez, 2010; Mansouritorghabeh, 2013) een bloeding beschreven. Hemostase werd beoordeeld als voldoende in 98% van de behandelingen (95% CI, 91-99%) (Scharrer, 1994; Rivard, 2008; Viswabandya, 2008). In een observationele studie met 13 patiënten was in 54% van de patiënten suppletie therapie postoperatief noodzakelijk (Venkataramani, 2000). Er werden geen trombotische events beschreven in de 3 studies (94 operaties) die deze uitkomstmaat bekeken (Franchi, 1995; Rivard, 2008; Viswabandya, 2008). In de case series waarbij alleen tranexaminezuur werd gegeven was er sprake van een bloeding in 14% van de ingrepen (95% CI, 9-20%) (Tavenner, 1972; Hewson, 2011; Davis, 2013; Lewandowski, 2018). Er werden geen significante adverse events beschreven.
Platelet type VWD en VWD type 2B
Bij platelet type VWD is er sprake van een afwijkend GPIb op de bloedplaatjes, waardoor er verhoogde affiniteit voor VWF ontstaat. Door deze spontane binding aan GPIb is er bij platelet type VWD vaak een trombocytopenie, die nog verder kan toenemen bij het geven van DDAVP of VWF/factor VIII concentraat. In 2020 publiceerde de Platelet Physiology Subcommittee van de ISTH een richtlijn voor de diagnostiek en management van platelet type VWD (Othman, 2020). Behandeling met VWF concentraat in combinatie met 1 HLA gematched trombocytenconcentraat wordt geadviseerd. Voor grote ingrepen wordt een VWF streefspiegel van 50-60 IE/dL geadviseerd en voor kleine ingrepen een VWF spiegel van 30-50 IE/dL.
Bij VWD type 2B kan er een trombocytopenie ontstaan door de verhooge affiniteit voor de GPIb receptor. Eventuele trombocytentransfusie in aanvulling op VWF(/FVIII) concentraat moet worden overwogen op basis van het trombocytenaantal. Voor de hoogte van de streeftrombocyten wordt verwezen naar de Bloedtransfusierichtlijn: Trombocytenwaarde voor profylactische trombocytentransfusie – Richtlijn – Richtlijnendatabase
Tromboserisico
In meerdere richtlijnen wordt geadviseerd om extreem hoge factor VIII spiegels te vermijden (> 150-300 IE/dL). In de studie van Koster et al. (Koster, 1995) bleek een factor VIII waarde > 150 IE/dL een onafhankelijke risicofactor te zijn voor het ontwikkelen van een diep veneuze trombose met een odds ratio (OR) van 4.8 (95%CI: 2.3-10.0). Een systematische review uit 2012 (Coppola, 2012) bekeek trombotische complicaties gerelateerd aan factor substitutie in patiënten met hemofilie en VWD in prospectieve studies. Er werden 71 studies geïncludeerd met in totaal 5528 patiënten met hemofilie A, hemofilie B en VWD. In totaal werden er 20 trombotische adverse events gerapporteerd (2 hemofilie A, 11 hemofilie B, 7 VWD). In de groep VWD patiënten werd, naast 5 oppervlakkige tromboflebitiden, 1 diep veneuze trombose (DVT) en 1 longembolie beschreven bij patiënten die langdurig VWF(/factor VIII) concentraat kregen in verband met een operatie en waarbij er sprake was van zeer hoge factor VIII waarden (patiënt met DVT: factor VIII 248 IE/dL, patiënt met longembolie: factor VIII 450 IE/dL).
Referenties
Bewijskracht literatuur
Level 2-3, consistent
Zoeken en selecteren
Er werd voor deze uitgangsvraag gebruik gemaakt van in 2021 door de American Society of Hematology (ASH), International Society on Thrombosis and Haemostasis (ISTH), National Hemophilia Foundation (NHF), and World Federation of Hemophilia (WFH) opgestelde en gepubliceerde evidence-based richtlijn; de Britse richtlijn voor diagnostiek en management van VWD van de United Kindom Haemophilia Centre Doctors Organisation (UKHCDO) uit 2014; de Nordic Haemophilia Council’s guideline uit 2011; de richtlijn Diagnostiek en behandeling van hemofilie en aanverwante hemostasestoornissen van de Nederland Vereniging van Hemofiliebehandelaars (NVHB) uit 2009 en de VWD richtlijn van de Amerikaanse National Heart, Lung, and Blood Institute (NHLBI) uit 2008.
Zoekverantwoording
De zoekverantwoording is opgenomen in diverse richtlijnen en bijbehorende (online) supplementen (Nichols, 2008; NVHB, 2009; Lassila, 2011; Laffan, 2014; Connell, 2021)
Evidence tabellen
De evidence tabellen van de gebruikte richtlijnen zijn weergegeven in het EtD raamwerk van de ASH ISTH NHF WFH richtlijn (Connell, 2021), online beschikbaar; op de website van de British Committee for Standards in Haematology (Laffan, 2014); op de website van de Nordic Haemophilia Counsil (Lassila, 2011); op de website van de NHLBI richtlijn (Nichols, 2008).
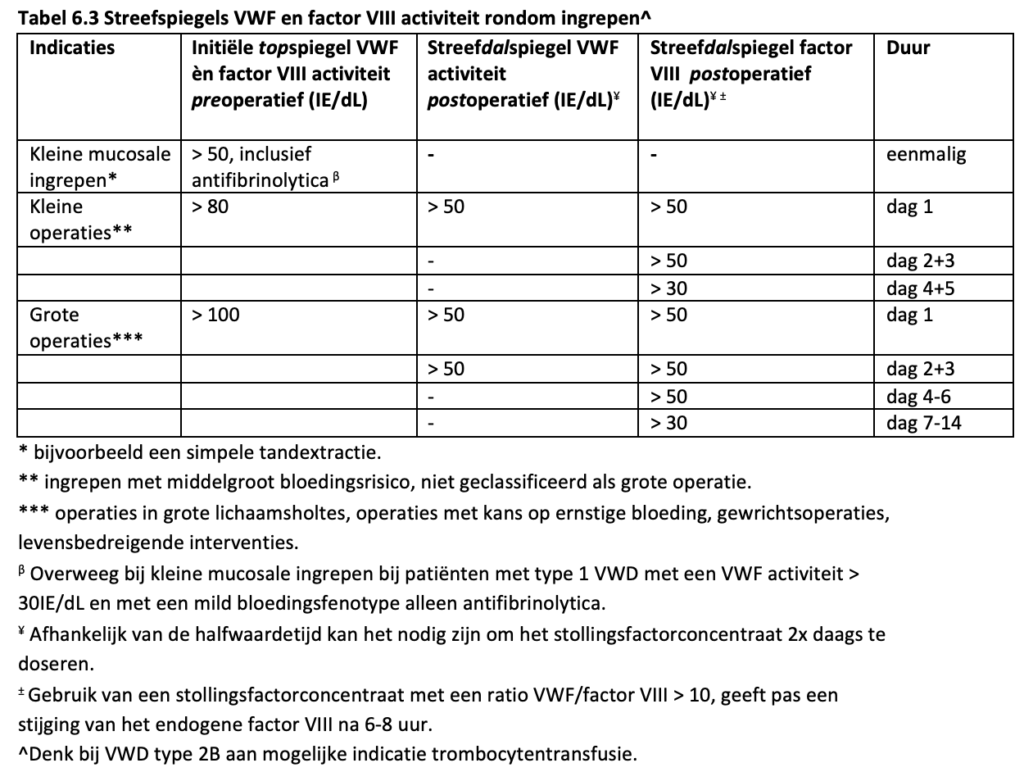
De in de studies gerapporteerde streefspiegels voor VWF en factor VIII rondom ingrepen bij VWD zijn weergegeven in de tabellen 6.1 (grote ingrepen) en 6.2 (kleine ingrepen).
Overwegingen
Streefspiegels VWF en factor VIII bij grote en kleine ingrepen
Er is veel variatie in de pre- en postoperatieve streefspiegels voor VWF en factor VIII en de duur van behandeling in de beschreven case series en richtlijnen. Over het algemeen werd bij grote ingrepen bijna altijd gekozen voor een preoperatieve VWF en/of factor VIII streefspiegel van 80-100 IE/dL en bij kleine ingrepen van > 50-80 IE/dL. De postoperatieve streefspiegels en duur van behandeling zijn zeer variabel. Bij grote ingrepen variërend van 50-100 IE/dL VWF en/of factor VIII in de eerste 7-10 dagen postoperatief, waarbij in de Noorse richtlijn ook nog op dag 7-14 een streefspiegel > 30 IE/dL geadviseerd werd (tabel 6.1). Voor kleine ingrepen varieerden de post operatieve streefspiegels van 30-50 IE/dL gedurende 1-7 dagen (tabel 6.2). In de studies werd hiermee in het overgrote deel van de ingrepen goede tot uitstekende hemostase bereikt. Er werden geen significante adverse events gerapporteerd. Wel werd duidelijk dat het doseren op basis van factor VIII leidde tot onnodig hoge VWF en factor VIII waarden.
Met het steeds meer beschikbaar komen van snelle VWF bepalingen, is het mogelijk om zowel de VWF als factor VIII peroperatief te meten en op basis van VWF en factor VIII streefspiegels een behandelplan te maken. De hoeveelheid stollingsfactorconcentraat kan berekend worden op basis van zowel de VWF concentratie en de factor VIII concentratie. Hierbij kan gebruik gemaakt worden van de onderstaande berekeningen.
Doordat de diverse VWF/factor VIII concentraten een verschillende verhouding VWF en factor VIII bevatten, dient er bij het doseren rekening te worden gehouden met wat de te verwachten stijging van zowel VWF activiteit als factor VIIII activiteit zal zijn op basis van de toegediende aantal eenheden per factor.
Tranexaminezuur bij kleine ingrepen
De case series lieten zien dat de synergistische werking van VWF en tranexaminezuur het risico op postoperatieve bloeding verlaagd. De aanbeveling om alleen tranexaminezuur te geven bij patiënten met type 1 VWD met spiegels > 30IE/dL en een mild bloedingsfenotype die kleine mucosale ingrepen moeten ondergaan, is met name gebaseerd op de angst op overbehandeling van deze patiënten en op de haalbaarheid van het voorschrijven van tranexaminezuur in een scenario waarin de kans op bloedingscomplicaties laag is en het vermijden van de last en de kosten van het toedienen van factorconcentraat bij deze patiënten. Daarnaast heeft tranexaminezuur een minimaal bijwerkingenprofiel.